19.3 — Monte Carlo methods
edits: — under construction —
Introduction
Statistical method that employ Monte Carlo methods use repeated random sampling to estimate properties of a frequency distribution. These distributions may be well-known, e.g., gamma-distribution, normal distribution, or t-distribution, or . The simulation is based on generation of a set of random numbers on the open interval (0,1) — the set of real numbers between zero and one (all numbers greater than 0 and less than 1).
If the set included 0 and 1, then it would be called a closed set, i.e., the set includes the boundary points zero and one.
Markov chain Monte Carlo (MCMC) sampling approach used to solve large scale problems. The Markov chain refers to how the sample is drawn from a specified probability distribution. It can be drawn by discrete time steps (DTMC) or by a continuous process (CTMC). The Markov process is “memoryless:” predictions of future events are derived solely from their present state — the future and past states are independent.
Gibbs sampling is a common MCMC algorithm.
ccc
R code
R’s uniform generator is runif function. Examples of the samples generated over different values (100, 1000, 10000, 100000) with output displayed as histograms (Fig. 1). Note that as sample size increases, the simulated distributions resemble more and more the uniform distribution. Use set.seed() to reproduce the same set and sequence of numbers
require(RcmdrMisc) par(mfrow = c(2, 2)) myUniformH <- data.frame(runif(100)) with(myUniformH, Hist(runif.100., scale="frequency", ylim=c(0,20), breaks="Sturges", col="red", xlab="100 samples", ylab="Count")) myUniform1K <- data.frame(runif(1000)) with(myUniform1K, Hist(runif.1000., scale="frequency", ylim=c(0,150), breaks="Sturges", col="green", xlab="1K samples", ylab="Count")) myUniform10K <- data.frame(runif(10000)) with(myUniform10K, Hist(runif.10000., scale="frequency", ylim=c(0,600), breaks="Sturges", col="lightblue", xlab="10K samples", ylab="Count")) myUniform100K <- data.frame(runif(100000)) with(myUniform100K, Hist(runif.100000., scale="frequency", ylim=c(0,5000),breaks="Sturges", col="blue", xlab="100K samples", ylab="Count")) #reset par() dev.off()
Yes, a nice repeating function would be more elegant code, but we move on. As a suggestion, you should create one! Use sapply() or a basic for loop.
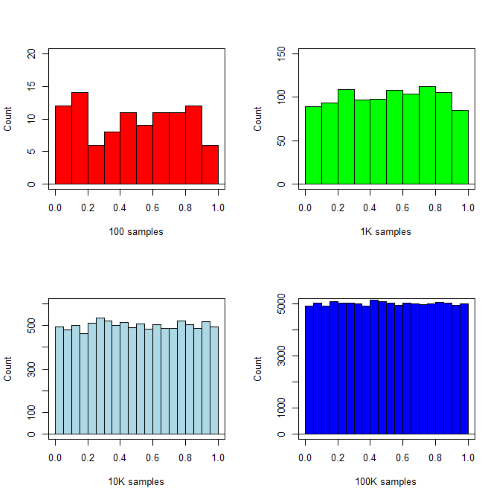
Figure 1. Histograms of runif results with 100, 1K, 10K, and 100K numbers of values to be generated
Looks pretty uniform. A property of random numbers is that history should not influence the future, i.e., no autocorrelation. We can check using the acf() function (Fig. 2).
par(mfrow = c(2, 2)) acf(myUniformH, main="100") acf(myUniform1K, main="1K") acf(myUniform10K, main="10K") acf(myUniform100K, main="100K" dev.off()
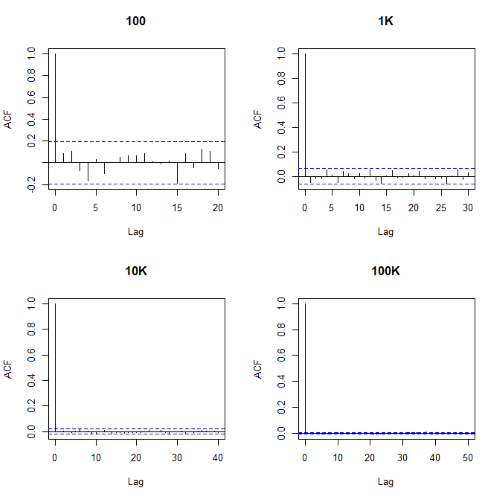
Figure 2. Autocorrelation plots of runif results with 100, 1K, 10K, and 100K numbers of values
Correlations among points are plotted versus lag, where lag refers to the number of points between adjacent points, e.g., lag = 10 reflects the correlation among points 1 and 11, 2 and 12, and so forth. The band defined by two parallel blue dashed lines
Questions
- Use
set.seed(123)and repeatrunif(10)twice. Confirm that the two sets are different (do not set seed) or the same whenset.seedis used. R hint: use functionidentical(x,y), where x and y are the two generated samples. This function tests whether the values and sequence of elements are the same between the two vectors.
Chapter 19 contents
- Introduction
- Jackknife sampling
- Bootstrap sampling
- Monte Carlo methods
- Ch19 References and suggested readings
16.3 – Data aggregation and correlation
Introduction
Correlations are easy to calculate, but interpretation beyond a strict statistical interpretation, eg, two variables linearly associated, may be complicated — caution is recommended. With respect to interpreting a correlation, caution and temperance is warranted. As previously discussed, “correlation is not causation,” is well known, but identifying when this applies to a particular analysis is not straight-forward. We introduced the problem of two variables sharing a hidden covariation which drives the correlation. In this section we introduce how correlations among grouped (aggregated) data may be quite different from the underlining individual correlations (cf Robertson 1950, Greenland 2001, Portnov et. al 2006).
Perfect (+1 or -1) correlation?
What if an estimated correlation value between two ratio scale variables turns out to be +1 or -1, the limits of the possible range of the correlation coefficient? Trivially, the reported value of one for the coefficient may be the result of rounding, it was actually 0.97. But, for argument’s sake, let’s assume the coefficient value was 1.0 to as many significant figures as the calculator may report. Another trivial possibility: suspect you’re looking at a case for which the two variables are the same thing, but on different scale. For example, the correlation between a range of temperatures measured in degrees Fahrenheit and then converted to degrees Celsius trivially will be +1.
Less trivially, a correlation of +1 or -1 may reveal that two variables are simply restatements of the same measured variable. Additionally, perfect correlation may reflect construction of a composite variable. Composite variables are examples of indexes, and are constructed from related variables by the researcher in order to better predict multivariate outcomes (see also Latent variables). Refs to add Diamantopoulos and Winklhofer 2001; Bollen and Diamantopoulos 2015
Data aggregation
Data aggregation or grouping refers to processes to group data in a summary form. Considerable public health data is presented this way. For example, CDC reports table after table of data about morbidity and mortality of United States of America population. Data are grouped by age, cities, counties, ethnicities, gender, and states and reports are generated to convey the status of health peoples. Similarly, education statistics, economic statistics, and statistics about crime are commonly crafted from grouped data of what originally was data for individuals.
Correlations between groups may yield spurious conclusions
Researchers interested in testing hypotheses like BMI is correlated with mortality (Flegal et al 2013, Kltasky et al 2017), or health disparities and ethnicity (Portnov et. al 2006), may use grouped data. In 16.2 we introduced the concept of spurious correlation. Correlations between grouped data may also mislead.
Consider the hypothesis that religiosity may deter criminal behavior. This hypothesis has been tested many times dating back to at least the 1940s (reviewed in Salvatore and Rubin 2018). Conclusions about religious beliefs range from negative association with criminal behavior to, in some reports, holding religious beliefs makes one more likely to commit crime. Testing versions of the hypothesis — what causes criminality in some individuals — among a variety of putative causal agents pops up through the history of biology research, arguably beginning with Galton. I hope you appreciate how challenging this would be to actually resolve — defining criminal behavior itself is laden with all kinds of sociology traps — and for a biologist, reeks of eugenics lore (Horgan 1993).
that all said, let’s proceed to test the religion-criminality hypothesis with aggregated data. The null hypothesis would be no association between crime statistics and numbers of churches. We can also ask about association between crime and non-religious or secular beliefs. I added numbers of Catholic churches and secular humanists groups for cities larger than 100K population by Internet search (FBI for crime statistics, Wikipedia for cities). Figure 1 and Figure 2 report crimes statistics aggregated by cities in the United States and by number of Catholic churches (Fig 1), and by number of secular humanists groups (Fig 2) in the same cities.
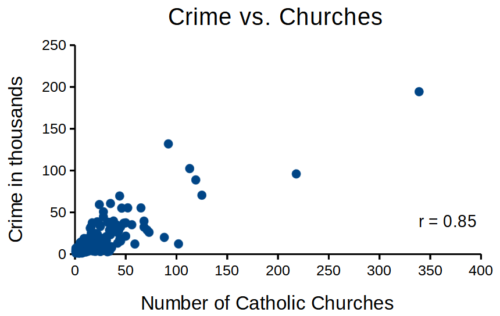
Figure 1. Scatterplot crime rates of cities by number of Catholic churches
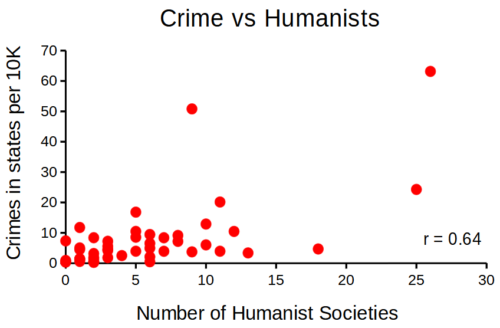
Figure 2. scatterplot crime rates of cities by number of secular humanist associations.
We’ll just take the numbers on faith (of course, we should think about the bunching around the origin — do we really think Internet search will get all of the secular groups, for example? Or is it really the case that several cities have no secular groups?). Both correlations were statistically different from zero, crime by churches (P < 0.001) and crime by secular groups (P < 0.001).
Now, having read Chapter 16.2 I trust you recognize immediately that there’s an important hidden covariate in common. Cities with small populations will have small numbers of crimes reported and smaller numbers of churches compared to large cities. Indeed, the correlation between population and crime for these cities was 0.89 and 0.97, respectively. However, after estimating the partial-correlations, we still have some explaining to do. For crime and churches, the partial correlation was +0.37 (p-value = 0.009); for crime and secular humanist groups, the partial correlation was -0.37 (p = 0.018). These results suggest that persons are more likely to commit crimes in cities with lots of Catholic churches whereas criminal behavior by individuals is less likely where secular humanist groups are numerous.
Before we start pointing fingers, the analysis presented here is a classic ecological fallacy. By grouping the data we lose information about the individuals, and it is the individuals to which the hypothesis applied. Thus, we are at risk of making incorrect conclusions by assuming that the individual is characterized by the group. The hypothesis remains challenging to test (how does one get a valid assessment of an individual’s religiosity? The hypothesis is challenging to test, but studies of individuals tend to find no association or a negative association between criminal behavior and religiosity (Salvatore and Rubin 2018). Crime statistics may underestimate criminal behavior, eg, embezzlement and other “white” crime), but a proper study would look to survey of individuals (Fig 3).
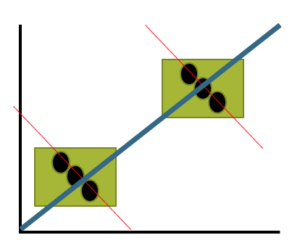
Figure 3. Illustration of ecological fallacy: positive association at level of groups (boxes, solid blue line), but negative association at level of individuals (black circles, red dashed lines).
Studies that use aggregate data test hypotheses about the groups, not about individuals in the groups. These studies are appropriate for comparing groups, eg, health disparities by ethnicity (Wang Kong et al 2022) or gender (Read and Gorman 2010; Cooper et al 2023), or comparisons among counties (eg, urban vs. rural) for medical resources (Anderson and Zimmerman 2024), or other “social determinates of health” (Crawfolrd 1977; Braveman and Gottlieb 2014), but one cannot conclude that the association is present for members of the group.
Bubble charts
Given we know about the covariation with population size, is there a better way to visualize the crime by number of churches and crime by number of humanist societies? One option is the bubble chart.
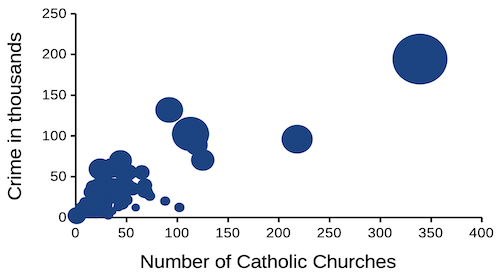
Figure 4. Bubble plot of data used to make Figure 1. Plot by LibreOffice Calc.
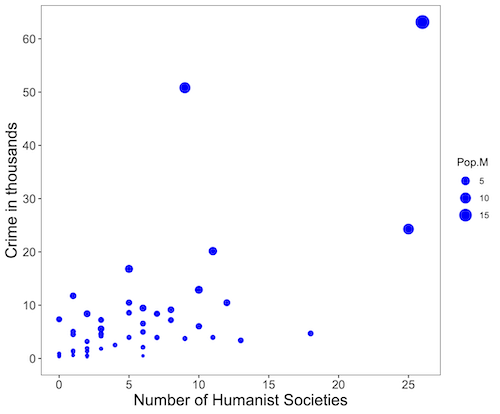
Figure 5. Bubble plot of data used to make Figure 2. Plot by ggplot2 package in R.
R code for Figure 5.
# STDHA site by Dr A. Kassambara https://www.sthda.com/english/
myPlot <- ggplot(humanists, aes(x = Humanist, y = Crime.K, size = Pop.M)) +
geom_point(color="blue", aes(size = Pop.M)) + theme_bw() + ylab("Crime in thousands") +
xlab("Number of Humanist Societies")
myPlot2 <- myPlot + theme(axis.title=element_text(size=16),
axis.text.x=element_text(size=12), axis.text.y=element_text(size=12),
panel.grid.major = element_blank(), panel.grid.minor = element_blank())
myPlot2 + scale_x_continuous(breaks=seq(0,30,5)) +
scale_y_continuous(breaks=seq(0,65,10))
Questions
1. Write up three learning outcomes for this page. Hint: Point your favorite generative AI to this page and ask for help.
2. What’s the most likely statistical explanation for why there is an apparent correlation between rates of autism and childhood vaccination rates? See Davidson 2017.
3. For the following data set, calculate the Pearson product moment correlation and, separately, the Spearman rank correlation. Report the values of the coefficients and provide an interpretation of the results.
v1: 68, 72, 68, 69, 65, 76, 58, 62, 69, 67, 66, 71, 70
v2: 1.727, 1.829, 1.727, 1.753, 1.651, 1.93, 1.473, 1.575, 1.753, 1.702, 1.676, 1.803, 1.778
4. For a small data set on human males ages 24 to 59, the estimated correlation between weight and the calculated BMI index was 0.68. (BMI is an example of a composite variable.) Explain why the correlation between weight and BMI is not +1.
Quiz Chapter 16.3
Data aggregation and correlation
Chapter 16 contents
18.3 – Logistic regression
Introduction
We briefly introduced logistic regression in the previous chapter on nonlinear regression. We expand our discussion of logistic regression here.
Logistic regression is a statistical method for modeling the dependence of a categorical (binomial) outcome variable on one or more categorical and continuous predictor variables (Bewick et al 2005).
The logistic function may be used to transform a sigmoidal curve to a more or less straight line while also changing the range of the data from binary (0 to 1) to infinity (-∞,+∞). For event with probability of occurring p, the logistic function is written as
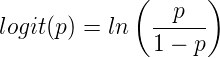
where ln refers to the natural logarithm.
This is an odds ratio. It represents the effect of the predictor variable on the chance that the event will occur.
The logistic regression model then very much resembles the same general linear models we have seen before.
![]()
In R and Rcmdr we use the glm() function to model the logistic function. Logistic regression is used to model a binary outcome variable. What is a binary outcome variable? It is categorical! Examples include: Living or Dead; Diabetes Yes or No; Coronary artery disease Yes or No. Male or Female. One of the categories could be scored 0, the other scored 1. For example, living might be 0 and dead might be scored as 1. (By the way, for a binomial variable, the mean for the variable is simply the number of experimental units with “1” divided by the total sample size.)
With the addition of a binary response variable, we are now really close to the Generalized Linear Model. Now we can handle statistical models in which our predictor variables are either categorical or ratio scale. All of the rules of crossed, balanced, nested, blocked designs still apply because our model is still of a linear form.
We write our generalized linear model

just to distinguish it from a general linear model with the ratio-scale Y as the response variable.
Think of the logistic regression as modeling a threshold of change between the 0 and the 1 value. In another way, think of all of the processes in nature in which there is a slow increase, followed by a rapid increase once a transition point is met, only to see the rate of change slow down again. Growth is like that (see Chapter 20.10 for related growth and related models). We start small, stay relatively small until birth, then as we reach our early teen years, a rapid change in growth (height, weight) is typically seed (well, not in my case … at least for the height). The curve I described is a logistic one (other models exist too). Where the linear regression function was used to minimize the squared residuals as the definition of the best fitting line, now we use the logistic as one possible way to describe or best fit this type of a curved relationship between an outcome and one or more predictor variables. We then set out to describe a model which captures when an event is unlikely to occur (the probability of dying is close to zero) AND to also describe when the event is highly likely to occur (the probability is close to one).
A simple way to view this is to think of time being the predictor (X) variable and risk of dying. If we’re talking about the lifetime of a mouse (lifespan typically about 18-36 months), then the risk of dying at one months is very low, and remains low through adulthood until the mouse begins the aging process. Here’s what the plot might look like, with the probability of dying at age X on the Y axis (probability = 0 to 1) (Fig 1).
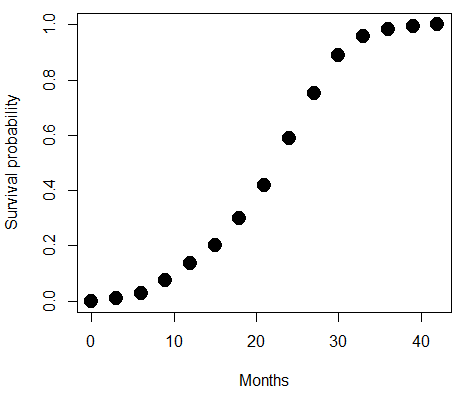
Figure 1. Lifespan of 1881 mice from 31 inbred strains (Data from Yuan et al [2012] available at https://phenome.jax.org/projects/Yuan2).
Note 1: I labeled Y axis labeled “Survival Probability”; “Inverse Survival Probability” would be more accurate.
We ask — of all the possible models we could draw — which model best fits the data? The curve fitting process is called the logistic regression. The sample data set is listed at end of this page (scroll down of click here). Create data.frame called yuan.
With some minor, but important differences, running the logistic regression is the same as what you have been doing so far for ANOVA and for linear regression. In Rcmdr, access the logistic regression function by calling the Generalized Linear Model (Fig 2).
Figure 2. Access Generalized Linear Model via R Commander
R results
GLM.1 <- glm(cumFreq ~ Months, family=gaussian(identity), data=yuan)
> summary(GLM.1)
Call:
glm(formula = cumFreq ~ Months, family = gaussian(identity),
data = yuan)
Deviance Residuals:
Min 1Q Median 3Q Max
-0.11070 -0.07799 -0.01728 0.06982 0.13345
Coefficients:
Estimate Std. Error t value Pr(>|t|)
(Intercept) -0.132709 0.045757 -2.90 0.0124 *
Months 0.029605 0.001854 15.97 6.37e-10 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
(Dispersion parameter for gaussian family taken to be 0.008663679)
Null deviance: 2.32129 on 14 degrees of freedom
Residual deviance: 0.11263 on 13 degrees of freedom
AIC: -24.808
Number of Fisher Scoring iterations: 2
Rcmdr: Statistics → Fit models → Generalized linear model.
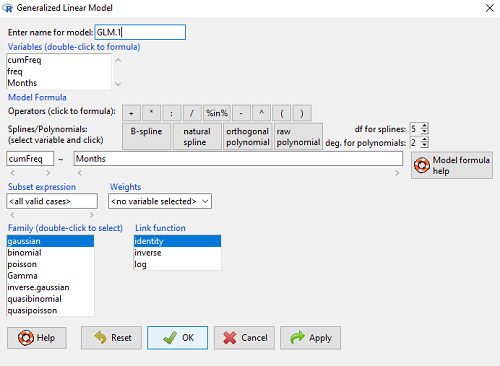
Figure 3. Screenshot Rcmdr GLM menu. For logistic on ratio-scale dependent variable, select gaussian family and identity link function.
Select the model as before. The box to the left accepts your binomial dependent variable; the box at right accepts your factors, your interactions, and your covariates. It permits you to inform R how to handle the factors: Crossed? Just enter the factors and follow each with a plus. If fully crossed, then the interactions may be specified with “:” to explicitly call for a two-way interaction between two (A:B) or a three-way interaction between three (A:B:C) variables. In the later case, if all of the two way interactions are of interest, simply typing A*B*C would have done it. If nested, then use %in% to specify the nesting factor.
R output
GLM.1 <- glm(cumFreq ~ Months, family=gaussian(identity), data=yuan)
summary(GLM.1)
Call:
glm(formula = cumFreq ~ Months, family = gaussian(identity),
data = yuan)
Deviance Residuals:
Min 1Q Median 3Q Max
-0.11070 -0.07799 -0.01728 0.06982 0.13345
Coefficients:
Estimate Std. Error t value Pr(>|t|)
(Intercept) -0.132709 0.045757 -2.90 0.0124 *
Months 0.029605 0.001854 15.97 6.37e-10 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
(Dispersion parameter for gaussian family taken to be 0.008663679)
Null deviance: 2.32129 on 14 degrees of freedom
Residual deviance: 0.11263 on 13 degrees of freedom
AIC: -24.808
Number of Fisher Scoring iterations: 2
Note 2: The %in% operator is an infix function used to determine if elements of a vector or object on the left-hand side are present within another vector or object on the right-hand side. Quietly, we introduced use of %in% in our code to subset data set in a BONUS problem.
Assessing fit of the logistic regression model
Some of the differences you will see with the logistic regression is the term “deviance.” deviance in statistics simply means compare one model to another and calculate some test statistic we’ll call “the deviance.” We then evaluate the size of the deviance like a chi-square goodness of fit. If the model fits the data poorly (residuals large relative to the predicted curve), then the deviance will be small and the probability will also be high — the model explains little of the data variation. On the other hand, if the deviance is large, then the probability will be small — the model explains the data, and the probability associated with the deviance will be small (significantly so? You guessed it! P < 0.05).
The Wald statistic
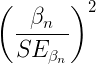
where n and β refers to any of the n coefficient from the logistic regression equation and SE refers to the standard error if the coefficient. The Wald test is used to test the statistical significance of the coefficients. It is distributed approximately as a chi-squared probability distribution with one degree of freedom. The Wald test is reasonable, but has been found to give values that are not possible for the parameter (eg, negative probability).
Likelihood ratio tests are generally preferred over the Wald test. For a coefficient, the likelihood test is written as
![]()
where L0 is the likelihood of the data when the coefficient is removed from the model (ie, set to zero value), whereas L1 is the likelihood of the data when the coefficient is the estimated value of the coefficient. It is also distributed approximately as a chi-squared probability distribution with one degree of freedom.
Nonlinear regression
Nonlinear regression, nls() function, may be a better choice. The following
attach(yuan)
logisticModel <-nls(cumFreq~DD/(1+exp(-(CC+bb*Months))), start=list(DD=1,CC=0.2,bb=.5),data=yuan,trace=TRUE)
summary(logisticModel)
Formula: yuan$cumFreq ~ DD/(1 + exp(-(CC + bb * yuan$Months)))
Parameters:
Estimate Std. Error t value Pr(>|t|)
DD 1.038504 0.014471 71.77 < 2e-16 ***
CC -4.626982 0.175109 -26.42 5.29e-12 ***
bb 0.206899 0.008777 23.57 2.03e-11 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Residual standard error: 0.01908 on 12 degrees of freedom
Number of iterations to convergence: 11
Achieved convergence tolerance: 0.000006909
Get fit statistics
AIC(logisticModel) [1] -71.54679
Because AIC for the nonlinear model much smaller (more negative) than AIC for logistic model, we may be tempted to judge fit of the nonlinear regression as best. However, this comparison of models is not valid because the Y variables are different between the two models and the fit families are different. One option, evaluate fit of models by plots of residuals (see 17.7 – Regression model fit).
Questions
1. Write up three learning outcomes for this page. Hint: Point your favorite generative AI to this page and ask for help.
Quiz Chapter 18.3
Logistic regression
Data set this page
| Months | freq | cumFreq |
|---|---|---|
| 0 | 0 | 0 |
| 3 | 0.010633 | 0.010633 |
| 6 | 0.017012 | 0.027645 |
| 9 | 0.045189 | 0.072834 |
| 12 | 0.064327 | 0.137161 |
| 15 | 0.064859 | 0.20202 |
| 18 | 0.09782 | 0.299841 |
| 21 | 0.118554 | 0.418394 |
| 24 | 0.171186 | 0.58958 |
| 27 | 0.162148 | 0.751728 |
| 30 | 0.137161 | 0.888889 |
| 33 | 0.069644 | 0.958533 |
| 36 | 0.024455 | 0.982988 |
| 39 | 0.011696 | 0.994684 |
| 42 | 0.005316 | 1 |
Chapter 18 contents
3.5 – Statistics of error
Making sense of error in data.
This page explains how statistics of error help quantify differences between measured and true values, beginning with classical methods like standard error and confidence intervals that rely on large-sample assumptions. Related, statisticians talk about uncertainty — the quantification of doubt about a measurement. We introduce permutation-based approaches, including jackknife and bootstrap resampling, which avoid those assumptions and are especially useful with smaller or complex data sets. Observer variation (intra- and interobserver error) is also highlighted as an important source of measurement error. We conclude with a discussion about confidence intervals as a measure of accuracy and emphasizes proper use of significant figures to avoid overstating precision.
Statistics of error.
In statistics, error doesn’t mean mistake, an avoidable human misjudgement or oversight, eg, a typo. An error in statistics means there was a difference between the measured value and the actual value for an object. Efforts by many authors of notable classical statistics developed a large body of calculated statistics, eg, standard error the mean, which allows the user to quantify how large the error of measurement is given assumptions about the distribution of the errors. Thus, classical statistics requires user to make assumptions about the error distribution, the subject of our Chapter 6. A critical issue to understand is that these methods assume large sample sizes are available; they are called asymptotic statistics or large-sample statistics; the properties of the statistical estimates are evaluated as sample size approaches infinity.
Jackknife sampling and bootstrap sampling are permutation approaches to working with data when the Central Limit Theorem — as sample size increases, the distribution of sample means will tend to a normal distribution (see Chapter 6.7) — is unlikely to apply or, rather, we don’t wish to make that assumption (Chapter 19). The jackknife is a sampling method involving repeatedly sampling from the original data set, but each time leaving one value out. The estimator, for example, the sample mean, is calculated for each sample. The repeated estimates from the jackknife approach yield many estimates which, collected, are used to calculate the sample variance. Jackknife estimators tend to be less biased than those from classical asymptotic statistics.
Bootstrapping, and not jackknife resampling, may now be the preferred permutation approach (eg, Google Scholar search “bootstrap statistics” 36K hits; “jackknife statistics” 17K hits), but which method is best depends on qualities of the data set. Bootstrapping involves large numbers of permutations of the original data, which, in short, means we repeatedly take many samples of our data and recalculate our statistics on these sets of sampled data. We obtain statistical significance by comparing our result from the original data against how often results from our permutations on the resampled data sets exceed the originally observed results. By permutation methods, the goal is to avoid the assumptions made by large-sample statistical inference, ie, reaching conclusions about the population based on samples from the population. Since its introduction, “bootstrapping” has been shown to be superior in many — but, importantly, not all — cases for statistics of error compared to the standard, classical approach (Efron 1979, Hesterberg 2015).
There are many advocates for the permutation approaches, and, because we have computers now instead of the hand calculators our statistics ancestors used, permutation methods may be the approach you will take in your own work. However, the classical approach has its strengths — when the conditions, that is, when the assumptions of asymptotic statistics are met by the data, then the classical approaches tend to be less conservative than the permutation methods. By conservative, statisticians mean that a test performs at the level we expect it to. Thus, if the assumptions of classical statistics are met they return the correct answer more often than do the permutation tests.
Error and the observer.
Individual researchers make observations, therefore, we can talk about observer variation as a kind of error measurement. For repeated measures of the same object by an individual, we would expect the individual to return the same results. To the extent repeated measures differ, this is intraobserver error. In contrast, measures of the same object from different individuals is interobserver error. For a new instrument or measurement system, one would need to establish the reliability of the measure: confronted with the same object do researchers get the same measurement? Accounting for interobserver error applies in many fields, eg, histopathology of putative carcinoma slides (Franc et al 2003), liver biopsies for cirrhosis (Rousselet et al 2005), blood cell counts (Bacus 1973).
Confidence in estimates.
A really useful concept in statistics is the idea that you can assign how confident you are to an estimate. This is another way to speak of the accuracy of an estimate. Clearly, we have more confidence in a sample estimate for a population parameter if many observations are made. Another factor in our ability to estimate is the magnitude of observation differences. In general, the larger the differences among values from repeated trials, the less confident we will be in out estimate, unless, again, we make our estimates from a large collection of observations. These two quantities, sample size and variability, along with our level of confidence, eg, 95%, are incorporated into a statistic called the confidence interval.

We will use this concept a lot throughout the course; for now, a simple but approximate confidence interval is to use the 2 x SEM rule (as long as sample size large): twice the standard error of the mean. Take your estimate of the mean, then add (upper limit) or subtract (lower limit) twice the value of the standard error of the mean (if you recall, that’s the standard deviation divided by the square-root of the sample size).
Example. Consider five magnetic darts thrown at a dart board (28 cm diameter, height of 1.68m from the floor) from a distance of 3.15 meters (Fig 1).

Figure 1. Magnetic dart board with 5 darts. Click image to view full sized image.
The distance in centimeters (cm) between where each of the five darts landed on the board compared to the bullseye is reported in (Table 1),
Table 1. Results of five darts thrown at a target (Figure 2)*
| Dart | Distance |
|---|---|
| 1 | 7.5 |
| 2 | 3.0 |
| 3 | 1.0 |
| 4 | 2.7 |
| 5 | 7.4 |
* distance in centimeters from the center.
Note 1: Use of the coordinate plane, and including the angle measurement in addition to distance (the vector) from center, would be a better analysis. In the context of darts, determining accuracy of a thrower is an Aim-Point targeting problem and part of your calculation would be to get MOA (minute of angle). For the record, the angles (degrees) were
| Dart | Distance | Angle |
|---|---|---|
| 1 | 7.5 | 124.4 |
| 2 | 3.0 | -123.7 |
| 3 | 1.0 | 96.3 |
| 4 | 2.7 | -84.3 |
| 5 | 7.4 | -31.5 |
measured using imageJ. Because there seems to be an R package for just about every data analysis scenario, unsurprisingly, there’s an R package called shotGroups to analyze shooting data.
How precise was the dart thrower? We’ll use the coefficient of variation as a measure of precision. Second, how accurate were the throws? Use R to calculate
darts = c(7.5, 3.0, 1.0, 2.7, 7.4) #use the coefficient of variation to describe precision of the throws coefVar = 100*(sd(darts)/mean(darts)); coefVar [1] 68.46141
Confidence Interval to describe accuracy.
For a game of darts, the true value would be a distance of zero — all bullseyes. We need to calculate the standard error of the mean (SEM), then, we calculate the confidence interval around the sample mean.
#Calculate the SEM SEM <- sd(darts)/sqrt(length(darts)); SEM [1] 1.322649 #now, get the lower and upper limit, subtract from the mean confidence <- c(mean(darts)-2*SEM, mean(darts), mean(darts)+2*SEM); confidence [1] 1.674702, 4.320000, 6.965298
The mean was 4.3 cm, therefore to get the lower limit of the interval subtract 2.65 (2*SEM = 2.645298) from the mean; for the upper limit add 2.65 to the mean. Thus, we report our approximate confidence interval as (1.7, 7.0), and we read this as saying we are about 95% confident the population value is between these two limits. Five is a very small sample number, so we shouldn’t be surprised to learn that our approximate confidence interval would be less than adequate. In statistical terms, we would use the t-distribution, and not the normal distribution, to make our confidence interval in cases like this.
Note 2: As a rule, implied by Central Limit theory and use of asymptotic statistical estimation, a sample size of 30 or more is safer, but probably unrealistic for many experiments. This is sometimes called as the rule of thirty. (For example, a 96-well PCR array costs about $500; with n = 30, that’s $15,000 US Dollars for one group!). So, what about this rule? This type of thinking should be avoided as “a relic of the pre-computer era,” (Hesterberg, T. (2008). It’s Time To Retire the” n>= 30″ rule.). We can improve on asymptotic statistics by applying bootstrap principles (Chapter 19).
We made a quick calculation of the confidence interval; we can get make this calculation by hand by incorporating the t distribution. We need to know the degrees of freedom, which in this case is 4 (n – 1, where n = 5). We look up critical value of t at 5% (to get our 95% confidence interval), t = 2.78. Subtract for lower limit t*SEM and add for upper limit t*SEM to the sample mean for the 95% confidence interval.
We can get help from R, by using the one-sample t-test with a test against the hypothesis that the true mean is equal to zero. Among a variety of output we will introduce later (see Chapter 8 and 10), the confidence interval is reported (yellow highlight),
#make an attach a data frame object Ddarts <- data.frame(darts) t.test(darts,mu=0) One Sample t-test data: darts t = 3.2662, df = 4, p-value = 0.0309 alternative hypothesis: true mean is not equal to 0 95 percent confidence interval: 0.6477381 7.9922619 sample estimates: mean of x 4.32
t.test uses the function qt(), which provides the quantile function. To recreate the 95% CI without the additional baggage output from the t.test, we would simply write
#upper limit
mean(darts)+ qt(0.975,df=4)*sd(darts)/sqrt(5)
#lower limit
mean(darts)+ qt(0.025, df = 4, lower.tail=TRUE)*sd(darts)/sqrt(5)
where sd(darts)/sqrt(5) is the standard error of the mean.
Or, alternatively, download and take advantage of a small package called Rmisc (not to be confused with the RcmdrMisc package) and use the function CI
library(Rmisc)
CI(darts)
upper mean lower
7.9922619 4.3200000 0.6477381
The advantage of using the CI() command from the package Rmisc is pretty clear; I don’t have to specify the degrees of freedom or the standard error of the mean. By default, CI reports the 95% confidence interval. we can specify any interval simply by adding to the command. For example,
CI(darts, ci=0.90)
reports upper and lower limits for the 90% confidence interval.
Try it. On your own, get the 90% confidence interval and the 99% confidence intervals for our dart example. Compare to the 95% confidence interval — what do you conclude about “confidence” in our estimate from 90% to 99%?
Confidence intervals from p-values.
And finally, it is not unusual for a report to provide summary statistics, differences between means (ie, effect size), and p-values (orange highlight), but not report confidence intervals. We discuss p-values and the concept of statistical significance beginning in Chapter 8. But for now, we show how to calculate approximate confidence intervals from the p-value. This may be of use for conducting meta-analysis (Chapter 20.15). See two helpful “how to” articles by Bland and Altman in 2011 BMJ (references listed in Chapter 3.6 – References).
In brief, calculate the test statistic for a normal distribution (letZ), calculate a standard error, and then the lower and upper limits for the 95% confidence interval. We illustrate use of the calculator formulas with the dart example, the p-value was 0.0309; the estimate was 4.32.
R code
letZ = (-0.862 + sqrt(0.743 - 2.404*log(0.0309, base = exp(1)))) = 2.154903
letSE = 4.32/letZ = 2.00473
1.96*letSE = 3.929271 # Add for upper and subtract for lower
The lower limit is 0.39 (4.32 – 3.929271) and the upper limit is 8.25 (4.32 + 3.929271). Although in the ballpark, these calculated estimates are wider than the confidence interval estimated by the t.test command. Small sample size, n = 5, is to blame — the accuracy of these methods, based on asymptotic statistical estimation approaches, assumes the law of large numbers applies.
Significant figures.
And finally, we should respect significant figures, the number of digits which have meaning. Our data were measured to the nearest tenth of a centimeter, or two significant figures. Therefore, if we report the confidence interval as (0.6477381, 7.9922619), then we imply a false level of precision, unless we also report our random sampling error of measurement.
R has a number of ways to manage output. One option would be to set number of figures globally with the options() function — all values reported by R would hold for the entire session. For example, options(digits=3) would report all numbers to three significant figures. Instead, I prefer to use signif() function, which allows us to report just the values we wish and does not change reporting behavior for the entire session.
signif(CI(darts),2) upper mean lower 8.00 4.30 0.65
Note 3: The options() function allows the R user to set a number of settings for an R session. After gaining familiarity with R, the advanced user recognizes that many settings can be changed to make the session work to report in ways more convenient to the user. If curious, submit options() at the R prompt and available settings will be displayed.
The R function signif() applies rounding rules. We apply rounding rules when required to report estimates to appropriate levels of precision. Rounding procedures are used to replace a number with a simplified approximation. Wikipedia provides a comprehensive list of rounding rules. Notable rules include
- directed rounding to an integer, eg, rounding up or down
- rounding to nearest integer, eg, round half up (down) if the number ends with 5
- randomly rounding to an integer, eg, stochastic rounding.
With the exception of stochastic rounding, all rounding methods impose biases on the sets of numbers. For example, the round half up method applied for numbers above 5, round down for numbers below 5 will increase the variance of the sample. In R, use round() for most of your work. If you need one of the other approaches, for example, to round up, the command is ceiling(); to round down we use floor().
When to round?
No doubt your previous math classes have cautioned you about the problems of rounding error and their influence on calculation. So, as a reminder, if reporting calls for rounding, then always round after you’ve completed your calculations, never during the calculations themselves.
A final note about significant figures and rounding. While the recommendations about reporting statistics are easy to come by (and often very proscriptive, eg, Table 1, Cole 2015), there are other concerns. Meta-analysis, which are done by collecting information from multiple studies, would benefit if more and not fewer numbers are reported, for the very same reason that we don’t round during calculations.
Questions
- Calculate the correct 90% and 99% confidence intervals for the dart data using the t-distribution
- by hand
- by one alternative method in R, demonstrated with examples in this page
- How many significant figures should be used for the volumetric pipettor p1000? The p200? The p20 (data at end of this page)?
- Another function,
round(), can be used. Try
round(CI(darts),2)
-
- and report the results: vary the significant figures from 1 to 10 (
signif()will take digits up to 22). - See any output differences between
signif()andround()? Don’t forget to take advantage of R help pages (eg, enter?roundat the R prompt) and see Wikipedia.
- and report the results: vary the significant figures from 1 to 10 (
- Compare rounding by
signif()andround()for the number 0.12345. Can you tell which rounding method the two functions use? - Calculate the coefficient of variation (CV) for each of the three volumetric pipettors from the data at end of this page. Rank the CV from smallest to largest. Which pipettor had the smallest CV and would therefore be judged the most precise?
- (data provided at end of this page, click here).
- Standards distinguish between within run precision and between run precision of a measurement instrument (see Chapter 16.5 – Instrument reliability and validity). The data in Table 1 were all recorded within 15 minutes by one operator. What kind pf precision was measured?
- Calculate the standard error of the means for each of the three pipettors from the data
- (data provided at end of this page, click here).
- Calculate the approximate confidence interval using the 2SE rule and judge which of the three pipettors is the most accurate (narrowest confidence interval)
- Repeat, but this time apply your preferred R method for obtaining confidence intervals.
- Compare approximate and R method confidence intervals. How well did the approximate method work?
- Return to the dart example, but now assume the p-value was p = 0.003 and again for p = 0.0003. Estimate the 95% confidence interval for each case; the difference remains 4.32. How does the change in p-value, from 0.03 to 0.0003 — a two orders of magnitude difference — affect the estimates for the confidence interval (hint: describe the width of the CI)?
Quiz Chapter 3.5
Statistics of error
Data sets
Pipette calibration
| p1000 | p200 | p100 |
|---|---|---|
| 0.113 | 0.1 | 0.101 |
| 0.114 | 0.1 | 0.1 |
| 0.113 | 0.1 | 0.1 |
| 0.115 | 0.099 | 0.101 |
| 0.113 | 0.1 | 0.101 |
| 0.112 | 0.1 | 0.1 |
| 0.113 | 0.1 | 0.1 |
| 0.111 | 0.1 | 0.1 |
| 0.114 | 0.101 | 0.101 |
| 0.112 | 0.1 | 0.1 |
Chapter 3 continues
19.2 – Bootstrap sampling
Introduction
Bootstrapping is a general approach to estimation or statistical inference that utilizes random sampling with replacement (Kulesa et al. 2015). In classic frequentist approach, a sample is drawn at random from the population and assumptions about the population distribution are made in order to conduct statistical inference. By resampling with replacement from the sample many times, the bootstrap samples can be viewed as if we drew from the population many times without invoking a theoretical distribution. A clear advantage of the bootstrap is that it allows estimation of confidence intervals without assuming a particular theoretical distribution and thus avoids the burden of repeating the experiment.
Base install of R includes the boot package. The boot package allows R users to work with most functions, and many authors have provided helpful packages. I highlight a couple packages
install packages lmboot, confintr
Example data set, Tadpoles from Chapter 14, copied to end of this page for convenience (scroll down or click here).
Bootstrapped 95% Confidence interval of population mean
Recall classic frequentist (large-sample) approach to confidence interval estimates of mean by R
x = round(mean(Tadpole$Body.mass),2); x
n = length(Tadpole$Body.mass); n
s = sd(Tadpole$Body.mass); s
error = qt(0.975,df=n-1)*(s/sqrt(n)); error
lower_ci = round(x-error,3)
upper_ci = round(x+error,3)
paste("95% CI of ", x, " between:", lower_ci, "&", upper_ci)
Output results are
> n = length(Tadpole$Body.mass); n [1] 13 > s = sd(Tadpole$Body.mass); s [1] 0.6366207 > error = qt(0.975,df=n-1)*(s/sqrt(n)); error [1] 0.384706 > paste("95% CI of ", x, " between:", lower_ci, "&", upper_ci) [1] "95% CI of 2.41 between: 2.025 & 2.795"
We used the t-distribution because both  the population mean and
the population mean and  the population standard deviation were unknown. Thus, 95 out of 100 confidence intervals would be expected to include the true value.
the population standard deviation were unknown. Thus, 95 out of 100 confidence intervals would be expected to include the true value.
Bootstrap equivalent
library(confintr)
ci_mean(Tadpole$Body.mass, type=c("bootstrap"), boot_type=c("stud"), R=999, probs=c(0.025, 0.975), seed=1)
Output results are
Two-sided 95% bootstrap confidence interval for the population mean based on 999 bootstrap replications and the student method Sample estimate: 2.412308 Confidence interval: 2.5% 97.5% 2.075808 2.880144
where stud is short for student t distribution (another common option is the percentile method — replace stud with perc), R = 999 directs the function to resample 999 times. We set seed=1 to initialize the pseudorandom number generator so that if we run the command again, we would get the same result. Any integer number can be used. For example, I set seed = 1 for output below
Confidence interval:
2.5% 97.5%
2.075808 2.880144
compared to repeated runs without initializing the pseudorandom number generator:
Confidence interval:
2.5% 97.5%
2.067558 2.934055
and again
Confidence interval:
2.5% 97.5%
2.067616 2.863158
Note that the classic confidence interval is narrower than the bootstrap estimate, in part because of the small sample size (i.e., not as accurate, does not actually achieve the nominal 95% coverage). Which to use? The sample size was small, just 13 tadpoles. Bootstrap samples were drawn from the original data set, thus it cannot make a small study more robust. The 999 samples can be thought as estimating the sampling distribution. If the assumptions of the t-distribution hold, then the classic approach would be preferred. For the Tadpole data set, Body.mass was approximately normally distributed (Anderson-Darling test = 0.21179, p-value = 0.8163). For cases where assumption of a particular distribution is unwarranted (e.g., what is the appropriate distribution when we compare medians among samples?), bootstrap may be preferred (and for small data sets, percentile bootstrap may be better). To complete the analysis, percentile bootstrap estimate of confidence interval are presented.
The R code
ci_mean(Tadpole$Body.mass, type=c("bootstrap"), boot_type=c("perc"), R=999, probs=c(0.025, 0.975), seed=1)
and the output
Two-sided 95% bootstrap confidence interval for the population mean based on 999 bootstrap replications
and the percent method
Sample estimate: 2.412308 Confidence interval: 2.5% 97.5% 2.076923 2.749231
In this case, the bootstrap percentile confidence interval is narrower than the frequentist approach.
Model coefficients by bootstrap
R code
Enter the model, then set B, the number of samples with replacement.
myBoot <- residual.boot(VO2~Body.mass, B = 1000, data = Tadpoles)
R returns two values:
bootEstParam, which are the bootstrap parameter estimates. Each column in the matrix lists the values for a coefficient. For this model,bootEstParam$[,1]is the intercept andbootEstParam$[,2]is the slope.origEstParam, a vector with the original parameter estimates for the model coefficients.seed, numerical value for the seed; use seed number to get reproducible results. If you don’t specify the seed, then seed is set to pick any random number.
While you can list the $bootEstParam, not advisable because it will be a list of 1000 numbers (the value set with B)!
Get necessary statistics and plots
#95% CI slope quantile(myBoot$bootEstParam[,2], probs=c(.025, .975))
R returns
2.5% 97.5%
335.0000 562.6228
#95% CI intercept quantile(myBoot$bootEstParam[,1], probs=c(.025, .975))
R returns
2.5% 97.5%
-881.3893 -310.8209
Slope
#plot the sampling distribution of the slope coefficient par(mar=c(5,5,5,5)) #setting margins to my preferred values hist(myBoot$bootEstParam[,2], col="blue", main="Bootstrap Sampling Distribution", xlab="Slope Estimate")
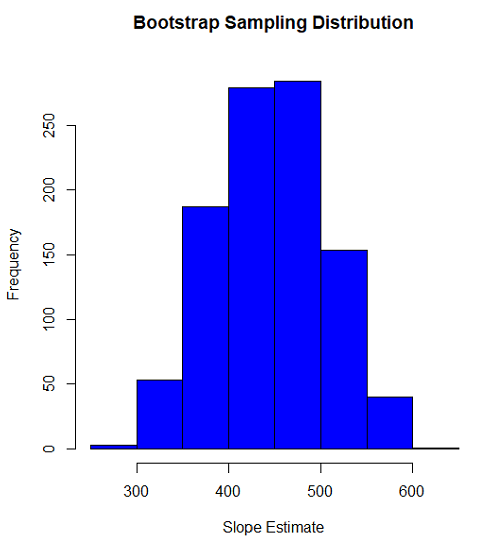
Figure 1. histogram of bootstrap estimates for slope
Intercept
#95% CI intercept quantile(myBoot$bootEstParam[,1], probs=c(.025, .975)) par(mar=c(5,5,5,5)) hist(myBoot$bootEstParam[,1], col="blue", main="Bootstrap Sampling Distribution", xlab="Intercept Estimate")
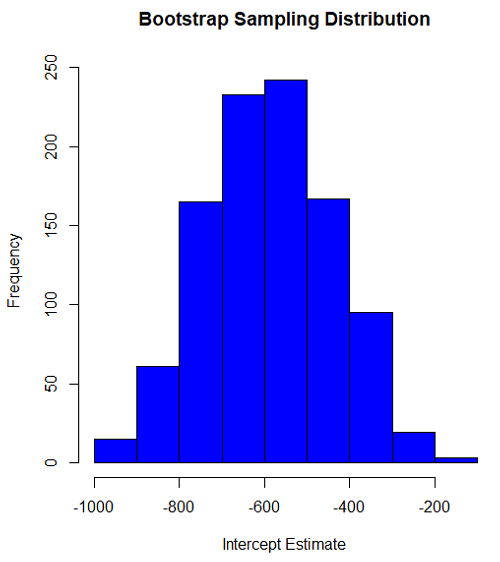
Figure 2. Histogram of bootstrap estimates for intercept.
Questions
edits: pending
Data set used this page (sorted)
| Gosner | Body mass | VO2 |
| I | 1.76 | 109.41 |
| I | 1.88 | 329.06 |
| I | 1.95 | 82.35 |
| I | 2.13 | 198 |
| I | 2.26 | 607.7 |
| II | 2.28 | 362.71 |
| II | 2.35 | 556.6 |
| II | 2.62 | 612.93 |
| II | 2.77 | 514.02 |
| II | 2.97 | 961.01 |
| II | 3.14 | 892.41 |
| II | 3.79 | 976.97 |
| NA | 1.46 | 170.91 |
Chapter 19 contents
- Introduction
- Jackknife sampling
- Bootstrap sampling
- Monte Carlo methods
- Ch19 References and suggested readings
19.1 – Jackknife sampling
Introduction
edits: — under construction —
R packages
There are several R packages one could use. The package bootstrap may be the the most general, and includes a jackknife routine suitable for any function. This page demonstrates jackknife estimate of correlation.
Example data set, cars, stopping distance by speed of car (scroll down or click here).
install package bootstrap
Jackknife estimates on linear models
These procedures can be done with the bootstrap package, but lmboot is a specific package to solve the problem
install package lmboot
Example data set, Tadpoles from Chapter 14, copied to end of this page for your convenience (scroll down or click here).
R code
jackknife(VO2~Body.mass, data = Tadpoles)
R returns two values:
bootEstParam, which are the jackknife parameter estimates. Each column in the matrix lists the values for a coefficient. For this model,bootEstParam$[,1]is the intercept andbootEstParam$[,2]is the slope.origEstParam, a vector with the original parameter estimates for the model coefficients.
$bootEstParam
(Intercept) Body.mass
[1,] -660.8403 472.6841
[2,] -539.5951 430.3990
[3,] -612.8495 454.5188
[4,] -512.5914 423.0815
[5,] -543.1577 434.2789
[6,] -572.3895 442.9176
[7,] -613.7873 451.2656
[8,] -594.0366 446.2571
[9,] -582.1833 443.5404
[10,] -598.2244 456.0599
[11,] -531.3152 415.2467
[12,] -555.7287 430.5604
[13,] -726.8522 512.1268
$origEstParam
[,1]
(Intercept) -583.0454
Body.mass 444.9512
Get necessary statistics and plots
#95% CI slope quantile(jack.model.1$bootEstParam[,2], probs=c(.025, .975))
R returns
2.5% 97.5% 417.5971 500.2940
#95% CI intercept quantile(jack.model.1$bootEstParam[,1], probs=c(.025, .975))
R returns
2.5% 97.5% -707.0486 -518.2085
Coefficient estimates
Slope
#plot the sampling distribution of the slope coefficient par(mar=c(5,5,5,5)) #setting margins to my preferred values hist(jack.model.1$bootEstParam[,2], col="blue", main="Jackknife Sampling Distribution", xlab="Slope Estimate")
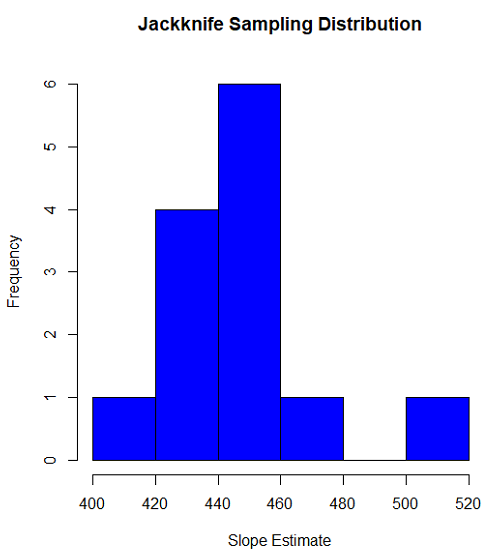
Figure 1. histogram of jackknife estimates for slope
Intercept
#95% CI intercept quantile(jack.model.1$bootEstParam[,1], probs=c(.025, .975)) par(mar=c(5,5,5,5)) hist(jack.model.1$bootEstParam[,1], col="blue", main="Jackknife Sampling Distribution", xlab="Intercept Estimate")
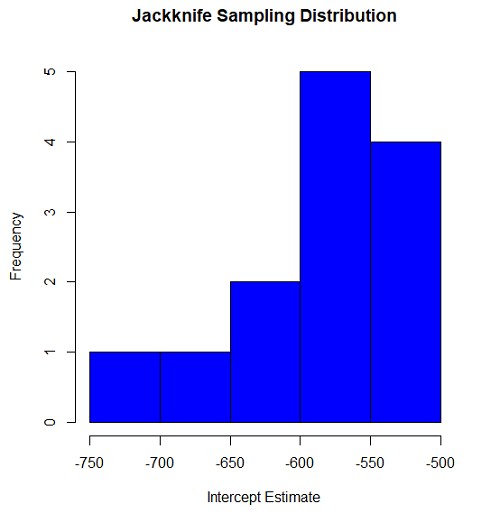
Figure 2. Histogram of jackknife estimates for intercept.
Questions
edits: pending
cars data set used this page
| speed | dist |
| 4 | 2 |
| 4 | 10 |
| 7 | 4 |
| 7 | 22 |
| 8 | 16 |
| 9 | 10 |
| 10 | 18 |
| 10 | 26 |
| 10 | 34 |
| 11 | 17 |
| 11 | 28 |
| 12 | 14 |
| 12 | 20 |
| 12 | 24 |
| 12 | 28 |
| 13 | 26 |
| 13 | 34 |
| 13 | 34 |
| 13 | 46 |
| 14 | 26 |
| 14 | 36 |
| 14 | 60 |
| 14 | 80 |
| 15 | 20 |
| 15 | 26 |
| 15 | 54 |
| 16 | 32 |
| 16 | 40 |
| 17 | 32 |
| 17 | 40 |
| 17 | 50 |
| 18 | 42 |
| 18 | 56 |
| 18 | 76 |
| 18 | 84 |
| 19 | 36 |
| 19 | 46 |
| 19 | 68 |
| 20 | 32 |
| 20 | 48 |
| 20 | 52 |
| 20 | 56 |
| 20 | 64 |
| 22 | 66 |
| 23 | 54 |
| 24 | 70 |
| 24 | 92 |
| 24 | 93 |
| 24 | 120 |
| 25 | 85 |
Data set used this page (sorted)
| Gosner | Body mass | VO2 |
| I | 1.76 | 109.41 |
| I | 1.88 | 329.06 |
| I | 1.95 | 82.35 |
| I | 2.13 | 198 |
| I | 2.26 | 607.7 |
| II | 2.28 | 362.71 |
| II | 2.35 | 556.6 |
| II | 2.62 | 612.93 |
| II | 2.77 | 514.02 |
| II | 2.97 | 961.01 |
| II | 3.14 | 892.41 |
| II | 3.79 | 976.97 |
| NA | 1.46 | 170.91 |
Chapter 19 contents
- Introduction
- Jackknife sampling
- Bootstrap sampling
- Ch19 References and suggested readings
17.6 – ANCOVA – analysis of covariance
Introduction
Analysis of covariance (ANCOVA) is intended to help with analysis of designs with categorical treatment variables on some response (dependent) variable, but a known confounding variable is also present. Thus, the researcher is also likely to know of additional ratio scale variables that covary with the response variable and, moreover, must be included in the experimental design in some way.
Take for example the well-known relationship between body size and whole-animal metabolic rate as measured by rates of carbon dioxide production or rates of oxygen consumption for aerobic organisms. We may be interested in how addition or blocking of stress hormones affects resting metabolism; we may be interested in comparing men and women for activity metabolism, and so on. We’d like to know if the regressions were the same (eg, metabolic rate covaried with body mass in the same way — that is, the slope of the relationship was the same).
This situation arises frequently in biology. For example, we might want to know if male and female birds have different mean field metabolic rates, in which case we might be tempted to use a one-way ANOVA or t-test (since one factor with two levels). However, if males and females also differ for body size, then any differences we might see in metabolic rate could be due to differences in metabolic rate are confounded by differences in the covariable body size. We already discussed one approach to correction: calculate a ratio. Thus, a logical approach to correcting or normalizing for the covariation would be to divide body mass (units of kilograms) into metabolic rate (eg, volume of oxygen, O2, consumed), and make comparisons, say, among different species, on mass-specific trait ( ). However, because the regression between mass and metabolic rate is allometric, i.e., not equal to one, the ratio does not, in fact normalize for body mass. We made this point in Chapter 6.2, and remarked that analysis of covariance ANCOVA was a solution.
). However, because the regression between mass and metabolic rate is allometric, i.e., not equal to one, the ratio does not, in fact normalize for body mass. We made this point in Chapter 6.2, and remarked that analysis of covariance ANCOVA was a solution.
ANCOVA allows you to test for mean differences in traits like metabolic rate between two or more groups, but only after first accounting for covariation due to another variable (eg, body size). However, ANCOVA makes the assumption that relationship between the covariable and the response variable is the same in the two groups. This is the same as saying that the regression slopes are the same. We discussed how to use t-test to test hypothesis of equal slopes between regression models in Chapter 17.5, but a more elegant way is to include this in your model.
Example
We return to our sample of 13 tadpoles (Rana pipiens), hatched in the laboratory. I’ve repeated the data set in this page, scroll or click here.
Our linear model was  and a scatterplot of the data set is shown in Figure 1 (a repeat of Figure 3 from Chapter 17.5, but now points identified to developmental group).
and a scatterplot of the data set is shown in Figure 1 (a repeat of Figure 3 from Chapter 17.5, but now points identified to developmental group).
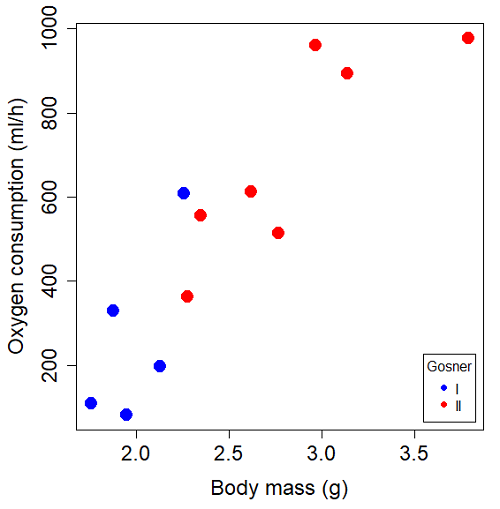
Figure 1. Scatterplot of oxygen consumption by R. pipiens tadpoles vs body mass (g) by developmental group (Gosner stages I or II).
The project looked at whether metabolism as measured by oxygen consumption was consistent across two developmental stages. Metamorphosis in frogs and other amphibians represents profound reorganization of the organism as the tadpole moves from water to air. Thus, we would predict some cost as evidenced by change in metabolism associated with later stages of development. Figure 2 shows a box plot of tadpole oxygen consumption by Gosner (1960) developmental stage (Figure 2 is a repeat of Figure 4 Chapter 17.5).
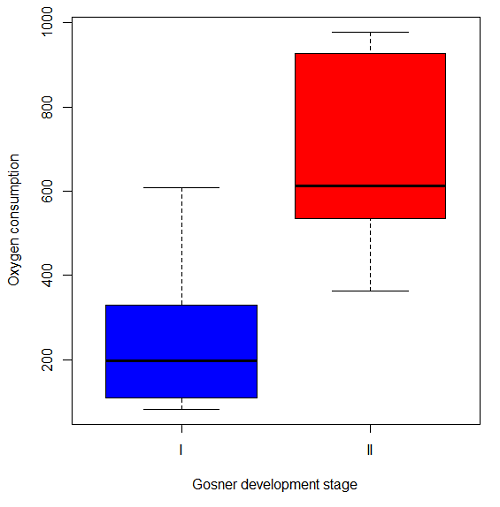
Figure 2. Copy of Figure 4, Chapter 17.5; boxplot of oxygen consumption of R. pipiens tadpoles by Gosner developmental stages.
Looking at Figure 2 we see a trend consistent with our prediction; developmental stage may be associated with increased metabolism. However, older tadpoles also tend to be larger, and the plot in Figure 2 does not account for that. Thus, body mass is a confounding variable in this example. There are several options for analysis here (eg, ANCOVA), but one way to view this is to compare the slopes for the two developmental stages. While this test does not compare the means, it does ask a related question: is there evidence of change in rate of oxygen consumption relative to body size between the two developmental stages? The assumption that the slopes are equal is a necessary step for conducting the ANCOVA.
So, divide the data set (Table 1) into two groups by developmental stage (12 tadpoles could be assigned to one of two developmental stages; one was at a lower Gosner stage than the others and so is dropped from the subset.
Table 1.  and
and  of 12 R. pipiens tadpole larvae by Gosner developmental stage.
of 12 R. pipiens tadpole larvae by Gosner developmental stage.
Gosner stage I
|
|
| 1.76 | 109.41 |
| 1.88 | 329.06 |
| 1.95 | 82.35 |
| 2.13 | 198 |
| 2.26 | 607.7 |
Gosner stage II
| Body mass | |
| 2.28 | 362.71 |
| 2.35 | 556.6 |
| 2.62 | 612.93 |
| 2.77 | 514.02 |
| 2.97 | 961.01 |
| 3.14 | 892.41 |
| 3.79 | 976.97 |
The slopes and standard errors we obtained in Chapter 17.5 were
| Gosner Stage I | Gosner stage II | |
| slope | 750.0 | 399.9 |
| standard error of slope | 444.6 | 111.2 |
Make a plot (Figure 3).
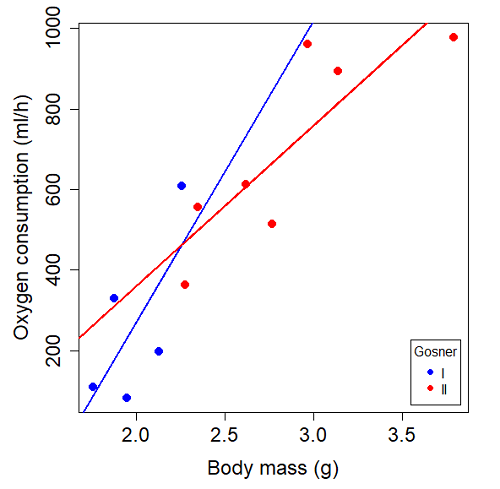
Figure 3. Scatterplot with best-fit regression lines of by for Gosner State I (closed circle, solid line) and Gosner Stage II (open circle, dashed line) R. pipiens tadpoles.
R code for plot in Figure 3.
#Used Rcmdr scatterplot(), then modified code
scatterplot(VO2~Body.mass | Gosner, regLine=FALSE, smooth=FALSE,
boxplots=FALSE, xlab="Body mass (g)", ylab="Oxygen consumption (ml/h)",
main="", cex=1.4, cex.axis=1.5, cex.lab=1.5, pch=c(19,19), by.groups=TRUE,
col=c("blue","red"), grid=FALSE,
legend=list(coords="bottomright"), data=Tadpoles)
#Get regression equations for groups, subset by Gosner
abline(lm(VO2~Body.mass, data=Stage01), lty=1, lwd=2, col="blue")
abline(lm(VO2~Body.mass, data=Stage02), lty=1, lwd=2, col="red")
Returning to the important question, are the two slopes statistically indistinguishable (Ho: bI = bII), where bI is the slope for the Gosner Stage I subset and bII is the slope for the Gosner Stage II subset? We look at the plot, and since the lines cross, we tend to see a difference. Of course, we need to consider that our perception of slope differences may simply be chance, especially because the sample size is small. Proceed to test.
R code
The ANCOVA is a new ANOVA model where the factor variables are adjusted or corrected for the effects of the continuous variable.
R code for ANCOVA example, crossed or interaction model.
tadpole.1 <- lm(VO2 ~ Body.mass*Gosner, data=example.Tadpole) summary(tadpole.1) Anova(tadpole.1, type="II")
Output
summary(tadpole.1)
Call:
lm(formula = VO2 ~ Body.mass * Gosner, data = example.Tadpole)
Residuals:
Min 1Q Median 3Q Max
-167.80 -117.93 13.81 94.66 214.65
Coefficients:
Estimate Std. Error t value Pr(>|t|)
(Intercept) -1231.6 783.0 -1.573 0.1544
Body.mass 750.0 390.7 1.919 0.0912 .
Gosner[T.II] 790.4 859.2 0.920 0.3845
Body.mass:Gosner[T.II] -350.1 409.5 -0.855 0.4174
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Residual standard error: 155.8 on 8 degrees of freedom
(1 observation deleted due to missingness)
Multiple R-squared: 0.821, Adjusted R-squared: 0.7539
F-statistic: 12.23 on 3 and 8 DF, p-value: 0.002336
The coefficients for the first factor (GII) and then the differences in the coefficient for the second factor. You can just add the second coefficient to the first so they’re on the same scale.
Anova Table (Type II tests)
Response: VO2
Sum Sq Df F value Pr(>F)
Body.mass 330046 1 13.6030 0.006146 **
Gosner 5630 1 0.2321 0.642908
Body.mass:Gosner 17736 1 0.7310 0.417423
Residuals 194102 8
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Suggests interaction is not significant, i.e., the slopes are not different.
We can then proceed to check to see if the intercepts are different, now that we’ve confirmed no significant difference in slope.
R code for ANCOVA as additive model
tadpole.2 <- lm(VO2 ~ Body.mass + Gosner, data=example.Tadpole) summary(tadpole.2) Anova(tadpole.2, type="II")
Output
> summary(tadpole.2)
Call:
lm(formula = VO2 ~ Body.mass + Gosner, data = example.Tadpole)
Residuals:
Min 1Q Median 3Q Max
-163.12 -125.53 -20.27 83.71 228.56
Coefficients:
Estimate Std. Error t value Pr(>|t|)
(Intercept) -595.37 239.87 -2.482 0.03487 *
Body.mass 431.20 115.15 3.745 0.00459 **
Gosner[T.II] 64.96 132.83 0.489 0.63648
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Residual standard error: 153.4 on 9 degrees of freedom
(1 observation deleted due to missingness)
Multiple R-squared: 0.8047, Adjusted R-squared: 0.7613
F-statistic: 18.54 on 2 and 9 DF, p-value: 0.0006432
Anova(tadpole.2, type="II")
Anova Table (Type II tests)
Response: VO2
Sum Sq Df F value Pr(>F)
Body.mass 330046 1 14.0221 0.004593 **
Gosner 5630 1 0.2392 0.636482
Residuals 211839 9
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Note 1. There is no test of interaction in the added model. This model would be appropriate IF the slopes are equal.
Instead of the interaction, or as added, try a nested model, with body mass nested within stage.
tadpole.3 <- lm(VO2 ~ Body.mass/Gosner, data=example.Tadpole)
summary(tadpole.3)
Call:
lm(formula = VO2 ~ Body.mass/Gosner, data = example.Tadpole)
Residuals:
Min 1Q Median 3Q Max
-168.66 -131.14 -20.28 90.33 225.36
Coefficients:
Estimate Std. Error t value Pr(>|t|)
(Intercept) -575.10 319.51 -1.800 0.1054
Body.mass 423.65 162.50 2.607 0.0284 *
Body.mass:Gosner[T.II] 21.95 63.73 0.344 0.7384
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Residual standard error: 154.4 on 9 degrees of freedom
(1 observation deleted due to missingness)
Multiple R-squared: 0.8021, Adjusted R-squared: 0.7581
F-statistic: 18.24 on 2 and 9 DF, p-value: 0.0006823
Gets the true coefficient (nested lm() version).
The two test different hypotheses.
lm(VO2 ~ Body.mass * Gosner) tests whether or not the regression has a nonzero slope.
lm(VO2 ~ Body.mass */Gosner) test whether or not the slopes and intercepts from different factors are statistically significant.
Questions
- Write up three learning outcomes for this page. Hint: Point your favorite generative AI to this page and ask for help.
- An OLS approach was used for the analysis of tadpole oxygen consumption body mass. Consider the RMA approach — would that be a more appropriate regression model? Explain why or why not.
- Consider an experiment in which you plan to administer a treatment that has a carry-over effect. For example, Compare and contrast “crossed” and “nested” designs.
- True or False. The nested design option for the ANCOVA assumes the slopes for the two groups of tadpoles for the regression line of by are equal. Explain your choice.
- Metabolic rates like oxygen consumption over time are well-known examples of allometric relationships. That is, many biological variables (eg, is related as aMb, where M is body mass, slope b is scaling exponent), and best evaluated on log-log scale. Repeat the analysis above on log10-transformed and for
- crossed design (eg, tadpole.1 model)
- added design (eg, tadpole.2 model)
- nested design (eg, tadpole.3 model)
- Create the plot and add the fitted lines from crossed design to the plot.
Note 2. About log-transform of a variable. The most straight-forward tact is to create two new variables. For example,
lgVO2 <- log10(VO2)
Another option is to transform the variables within the call to lm() function. For example, try
lm(log10(VO2) ~ log10(Body.mass ), data=example.Tadpole)
Hint: don’t forget to attach your data set to avoid having to call the variable as, for example, example.Tadpole$VO2
Quiz Chapter 17.6
ANCOVA - analysis of covariance
Data sets
Oxygen consumption,  , of Anuran tadpoles, dataset=
, of Anuran tadpoles, dataset=example.Tadpole
| Gosner | Body mass | VO2 |
|---|---|---|
| NA | 1.46 | 170.91 |
| I | 1.76 | 109.41 |
| I | 1.88 | 329.06 |
| I | 1.95 | 82.35 |
| I | 2.13 | 198 |
| II | 2.28 | 362.71 |
| I | 2.26 | 607.7 |
| II | 2.35 | 556.6 |
| II | 2.62 | 612.93 |
| II | 2.77 | 514.02 |
| II | 2.97 | 961.01 |
| II | 3.14 | 892.41 |
Gosner refers to Gosner (1960), who developed a criteria for judging metamorphosis staging.
Chapter 17 contents
- Introduction
- Simple Linear Regression
- Relationship between the slope and the correlation
- Estimation of linear regression coefficients
- OLS, RMA, and smoothing functions
- Testing regression coefficients
- ANCOVA – Analysis of covariance
- Regression model fit
- Assumptions and model diagnostics for Simple Linear Regression
- References and suggested readings (Ch17 & 18)
18.6 – Compare two linear models
Introduction
Rcmdr (R) provides a very useful tool to compare models. Now, you can compare any two models, but this would be a poor strategy. Use this tool to perform in effect a stepwise test by hand. As one of the models, select for example the saturated model, then for the second model, select one in which you drop one model factor. In the example below, I dropped the two-way interaction from the saturated model (a logistic regression model, actually):
The model was Type.II diabetes = Treatment + Samples + Gender + BMI + Age + Gender:Treatment
where Type.II diabetes is a binomial (Yes,No) dependent variable and Treatment and Gender were categorical factors. The ANOVA table is shown below.
Anova(GLM.1, type="II", test="LR")
Analysis of Deviance Table (Type II tests)
Response: Type.II
LR Chisq Df Pr(>Chisq)
Treatment 0.266 7 0.9999
Samples 38.880 1 4.508e-10 ***
Gender 0.671 1 0.4127
BMI 2.259 1 0.1329
Age 2.064 1 0.1508
Treatment:Gender 1.803 1 0.1794
From this output we see that there are a number of terms that are not significant (P < 0.05), but with one exception (Treatment) they seem to contribute to the total variation (P values are between 0.13 and 0.4). So, we conclude that the saturated model is not the best fit model, and proceed to evaluate alternative models in search of the best one.
As a matter of practice I first drop the interaction term. Here’s the ANOVA table for the second model now without the interaction
Anova(GLM.1, type="II", test="LR")
Analysis of Deviance Table (Type II tests)
Response: Type.II
LR Chisq Df Pr(>Chisq)
Treatment 0.266 7 0.9999
Samples 37.086 1 1.13e-09 ***
Gender 0.671 1 0.4127
BMI 2.017 1 0.1556
Age 1.794 1 0.1804
Both models look about the same. Which one is best? We now wish to know if dropping the interaction harms the model in any way. We will use the AIC (Akaike Information Criterion) to evaluate the models. AIC provides a way to assess which among a set of nested models is better. The preferred model is the one with the lowest AIC value.
To access the AIC calculation, just enter the script AIC(model name), where model name refers to one of the models you wish to evaluate (e.g., GLM.1), then submit the code
AIC(GLM.1) 50.65518 AIC(GLM.2) 50.45793
Thus, we prefer the second model (GLM.2) because the AIC is lower.
AIC does not provide a statistical test of model fit. To access the model comparison tool, simply select
Models → Hypothesis tests → Compare two models…
and the following screen will appear (Fig 1).
Figure 1. Screenshot Rcmdr compare models menu.
Select the two models to compare (in this case, GLM1 and GLM2), then press OK button. R output
anova(GLM.1, GLM.2, test="Chisq")
Analysis of Deviance Table
Model 1: Type.II ~ Treatment + Samples + Gender + BMI + Age + Gender:Treatment
Model 2: Type.II ~ Treatment + Samples + Gender + BMI + Age
Resid. Df Resid. Dev Df Deviance P(>|Chi|)
1 49 24.655
2 50 26.458 -1 -1.8027 0.1794
We see that P >0.05 (= 0.1794), which means the fit of the model is fine if we lose the one term.
Deviance
Those of you working with logistic regressions will see this new term, “deviance.” Deviance is a statistical term relevant to model fitting. Think of it like a chi-square test statistic. The idea is that you compare your fitted model against the data in which the only thing estimate is the intercept. Do the additional components of the model add significantly to the prediction of the original data? If they do, dropping the term will have a significant effect on the model fit and the P-value would be less than 0.05. In this example, we see that dropping the interaction term had little effect on the deviance score and in agreement, the P value is larger than 0.05. It means we can drop the term and the new model lacking the term is in some sense better: fewer predictors, a simpler model.
Questions
- Write up three learning outcomes for this page. Hint: Point your favorite generative AI to this page and ask for help.
Quiz Chapter 18.6
Compare two linear models
Chapter 18 contents
18.5 – Selecting the best model
Introduction
This is a long entry in our textbook, many topics to cover. We discuss aspects of model fitting, from why model fitting is done to how to do it and what statistics are available to help us decide on the best model. Model selection very much depends on what the intent of the study is. For example, if the purpose of model building is to provide the best description of the data, then in general one should prefer the full (also called the saturated) model. On the other hand, if the purpose of model building is to make a predictive statistical model, then a reduced model may prove to be a better choice. The text here deals mostly with the later context of model selection, finding a justified reduced model.
From Full model to best Subset model
Model building is an essential part of being a scientist. As scientists, we seek models that explain as much of the variability about a phenomenon as possible, but yet remain simple enough to be of practical use.
Having just completed the introduction to multiple regression, we now move to the idea of how to pick best models.
We distinguish between a full model, which includes as many variables (predictors, factors) as the regression function can work with, returning interpretable, if not always statistically significant output, and a saturated model.
The saturated model is the one that includes all possible predictors, factors, interactions in your experiment. In well-behaved data sets, the full model and the saturated model will be the same model. However, they need not be the same model. For example, if two predictor variables are highly collinear, then you may return an error in regression fitting.
For those of you working with meta-analysis problems, you are unlikely to be able to run a saturated model because some level of a key factor are not available in all or at least most of the papers. Thus, in order to get the model to run, you start dropping factors, or you start nesting factors. If you were unable to get more things in the model, then this is your “full” model. Technically we wouldn’t call it saturated because there were other factors, they just didn’t have enough data to work with or they were essentially the same as something else in the model.
Identify the model that does run to completion as your full model and proceed to assess model fit criteria for that model, and all reduced models thereafter.
In R (Rcmdr) you know you have found the full model when the output lacks “NA” strings (missing values) in the output. Use the full model to report the values for each coefficient, i.e., conducting the inferential statistics.
Get the estimates directly from the output from running the regression function. You can tell if the effect is positive (look at the estimate for sample — it is positive) so you can say — more samples, greater likelihood to see more cases of cancer.
Remember, the experimental units are the papers themselves, so studies with larger numbers of subjects are going to find more cases of diabetes. We would worry big time with your project if we did not see statistically significant and positive for sample size.
Example: A multiple linear model
For illustration, here’s an example output following a run with the linear model function on a portion of the Kaggle Cardiovascular disease data set.
Note: For my training data set I simply took the first 100 rows — from the 70,000 rows! in the dataset. A more proper approach would include use of stratified random sampling, for example, by gender, alcohol use, and other categorical classifications.
The variables (and data types) were
SBP = systolic blood pressure (from ap_hi)
age.years = Independent variable, ratio scale (calculated from age in days)
BMI = Dependent variable, ratio scale (calculated from height and weight variables)
active = Independent variable, physical activity, binomial
alcohol = Independent variable, alcohol use, binomial
cholesterol = Independent variable, cholesterol levels (scored 1 = “normal,” 2 = “above normal, and 3 = “well above normal”)
gender = Independent variable, binomial
glucose = Independent variable, glucose levels (scored 1 = “normal,” 2 = “above normal, and 3 = “well above normal”)
smoke = Independent variable, binomial
R output
Call:
lm(formula = SBP ~ BMI + gender + age.years + active + alcohol +
cholesterol + gluc + smoke, data = kaggle.train)
Residuals:
Min 1Q Median 3Q Max
-33.359 -9.701 -1.065 8.703 45.242
Coefficients:
Estimate Std. Error t value Pr(>|t|)
(Intercept) 76.7664 14.9311 5.141 0.00000155 ***
BMI 0.8395 0.2933 2.862 0.00522 **
gender -2.0744 1.6102 -1.288 0.20092
age.years 0.2742 0.2403 1.141 0.25672
active 7.1411 3.4934 2.044 0.04383 *
alcohol -7.9272 9.1854 -0.863 0.39039
cholesterol 7.4539 2.3200 3.213 0.00182 **
gluc -1.2701 2.9533 -0.430 0.66817
smoke 11.4132 6.7015 1.703 0.09197 .
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Residual standard error: 14.96 on 91 degrees of freedom
Multiple R-squared: 0.2839, Adjusted R-squared: 0.221
F-statistic: 4.51 on 8 and 91 DF, p-value: 0.0001214
Question. Write out the equation in symbol form.
We see from the modest R2 (adjusted) that the model explains little variation in systolic blood pressure; only BMI, physical activity (yes, no), and serum cholesterol levels, were statistically significant at 5% level. Smoker (yes, no) approached significance (p-value = 0.092), but I wouldn’t go on and on about positive or negative coefficients and effects.
Note: For my training data set I simply took the first 100 rows — from the 70,000 rows! in the dataset. A more proper approach would include use of stratified random sampling, for example, by gender, alcohol use, and other categorical classifications.
We report the value (I would round to 7.5) for cholesterol on SBP (Table 1), and note that those who had elevated serum cholesterol levels tended to have higher systolic blood pressure (e.g., the sign of the coefficient — and I related the coefficient back to the most important thing about your study — the biological interpretation). Repeat the same for the other significant coefficients.
Now’s a good time to be clear about HOW you report statistical results. DO NOT SIMPLY COPY AND PASTE EVERYTHING into your report. Now, for the estimates above, you would report everything, but not all of the figures. Here’s how the output should be reported in your Project paper.
Table 1. Coefficients from full model.
term Coefficient Error P-value* Intercept 76.8 14.93 < 0.00001 BMI 0.8 0.29 0.00522 gender -2.1 1.61 0.20092 age.years 0.3 0.24 0.25672 active 7.1 3.49 0.04383 alcohol -7.9 9.19 0.39039 cholesterol 7.5 2.32 0.00182 gluc -1.3 2.95 0.66817 smoke 11.4 6.70 0.09197
* t-test
Looks better, doesn’t it?
Note: Obviously, don’t round for calculations!
Once you have the full model, we should run simple diagnostics on the regression fit to the data (VIF, Cook’s Distance, basic diagnostic plots), then proceed to use this model for the inferential statistics.
> vif(model.1) BMI gender age.years active alcohol cholesterol gluc smoke 1.112549 1.091154 1.081641 1.048519 1.096382 1.178635 1.168442 1.131081
No VIF was greater than 2, suggesting little multicollinearity among the predictor variables.
> cooks.distance(model.1)[which.max(cooks.distance(model.1))] 60 0.355763
This person had high SBP (180), was a smoker, but active and normal BMI.
Use the significance tests of each parameter in the model from the corresponding ANOVA table. Now, where is the ANOVA table? Remember, right after running the linear regression,
Rcmdr: Models → Hypothesis testing → ANOVA tables
Accept the default (partial marginality), and, Boom! … the ANOVA table you should be familiar with.
From the ANOVA table you will tell me whether a Factor is significant or not. You report the ANOVA table in your paper. You describe it.
Now, the next step is to decide what is the best model. It then guides you to the next step which is to decide whether a better model (fewer parameters, Occam’s razor) can be found. Identify the parameter from the ANOVA table with the highest P-value and remove it from the model when you run the regression again. Repeat the steps above, return the ANOVA table, checking the estimates and P-values, until you have a model with only statistically significant parameters.
Find the best model
We may be tempted to run the linear model again, but with non-significant predictors excluded. For example, re-run the model on SBP but with just BMI, active, cholesterol and excluding smoke.
We can calculate the AIC, Akaike Information Criterion, the lower value is considered the better model.
AIC(model.1) [1] 835.4938
AIC(model.2) [1] 834.0782
The full model had eight predictor variables, excluding the intercept, whereas the reduced model had just three predictor variables, again, not including the intercept. Unsurprisingly, the reduced model is better. But now, a stronger test — what about smoking, where the p-value approached 5%? We ran the linear model (model.3).
> AIC(model.3)
[1] 831.9495
We can use an anova test or the likelihood ratio test to compare the models. The anova test, the models must be nested.
stats::anova(model.2, model.3) Analysis of Variance Table Model 1: SBP ~ BMI + active + cholesterol Model 2: SBP ~ BMI + active + cholesterol + smoke Res.Df RSS Df Sum of Sq F Pr(>F) 1 96 22205 2 95 21307 1 898.1 4.0043 0.04824 * --- Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
and
lmtest::lrtest((model.2,model.3) Likelihood ratio test Model 1: SBP ~ BMI + active + cholesterol Model 2: SBP ~ BMI + active + cholesterol + smoke #Df LogLik Df Chisq Pr(>Chisq) 1 5 -412.04 2 6 -409.97 1 4.1286 0.04216 * --- Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Both tests tell us the same thing — our model with four predictors, including smoking, is better.
2 December 2024 — Past this point, edits pending
But first, I want to take up an important point about your models that you may not have had a chance to think about. The order of entry of parameters in your model can effect the significance and value of the estimates themselves. The order of parameter model entry above can be read top to bottom. Age was first, followed in sequence by CalsPDay, CholPDay, and so on. By convention, enter the covariates first (the ratio-scale predictors), that’s what I did above.
Here’s the output from a model in which I used a different order of parameters.
Anova(LinearModel.2, type="II")
Anova Table (Type II tests)
Response: BMI
Sum Sq Df F value Pr(>F)
Sex 1.52 1 0.0478 0.82764
Smoke 8.84 1 0.2782 0.59964
Age 0.62 1 0.0196 0.88918
CalsPDay 10.79 1 0.3394 0.56215
CholPDay 92.37 1 2.9065 0.09286 .
Sex:Smoke 3.35 1 0.1055 0.74631
Residuals 2129.34 67
The output is the same!!! So why did I give you a warning about parameter order? Run the ANOVA table summary command again, but this time select Type III type of test, i.e., ignore marginality
> Anova(LinearModel.2, type="III")
Anova Table (Type III tests)
Response: BMI
Sum Sq Df F value Pr(>F)
(Intercept) 1544.34 1 48.5929 1.708e-09 ***
Sex 4.69 1 0.1475 0.70214
Smoke 11.82 1 0.3720 0.54400
Age 0.62 1 0.0196 0.88918
CalsPDay 10.79 1 0.3394 0.56215
CholPDay 92.37 1 2.9065 0.09286 .
Sex:Smoke 3.35 1 0.1055 0.74631
Residuals 2129.34 67
The output has changed — and in fact it now reports the significance test of the intercept. This output is the same as the output from the linear model. Try again, this time selecting Type I, sequential
> anova(LinearModel.2)
Analysis of Variance Table
Response: BMI
Df Sum Sq Mean Sq F value Pr(>F)
Sex 1 0.68 0.681 0.0214 0.88402
Smoke 1 2.82 2.816 0.0886 0.76690
Age 1 3.44 3.436 0.1081 0.74333
CalsPDay 1 2.27 2.272 0.0715 0.78998
CholPDay 1 96.30 96.299 3.0301 0.08633
Sex:Smoke 1 3.35 3.354 0.1055 0.74631
Residuals 67 2129.34 31.781
Here, we see the effect of order. So, as we are working to learn all of the issues of statistics and in particular mode fitting, I have purposefully restricted you to Type II analyses — obeying marginality correctly handles most issues about order of entry.
Easy there…. Take a deep breath, and guess what? Your best model needs to have significant parameters in it, right? Your best fit model then is Model 5. And that model will be your candidate for best fit as we proceed to complete our model building.
![]()
Now we proceed to gain some support evidence for our candidate best model. We are going to use an information criterion approach.
Use a fit criterion for determining model fit
To help us evaluate evidence in favor of one model over another there are a number of statistics one may calculate to provide a single number for each model for comparison purposes. The criteria model evaluators available to us include Mallow’s Cp, adjusted R2, Akaike Information Criterion (AIC) or the Bayesian Information Criterion (BIC) to select best model.
We already introduced the coefficient of determination R2 as a measure of fit – in general we favor models with larger values of R2. However, values of R2 will always be larger for models with more parameters. Thus, the other evaluators attempt to adjust for the parameters in the model and how they contribute to increased model fit. For illustrative purposes we will use Mallow’s Cp. The equation for Mallow’s Cp in linear regression is
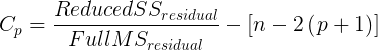
where p is the number of parameters in the model. Mallow’s Cp is thus equal to the number of parameters in the model plus an additional amount due to lack of fit of the model (i.e., large residuals). All else being equal we favor the model in which the Cp is close to the number of parameters in the model.
In Rcmdr, select Models → Subset model selection … (Fig. 1)
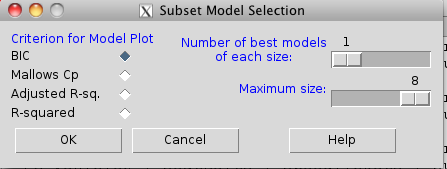
Figure 1. Rcmdr popup menu, Subset model selection…
From the menu, select the criterion and how many models to return. The function returns a graph that can be used to interpret which model is best given the selection criterion used. Below is an example (although for a different data set!) for Mallow’s Cp (Fig. 2).
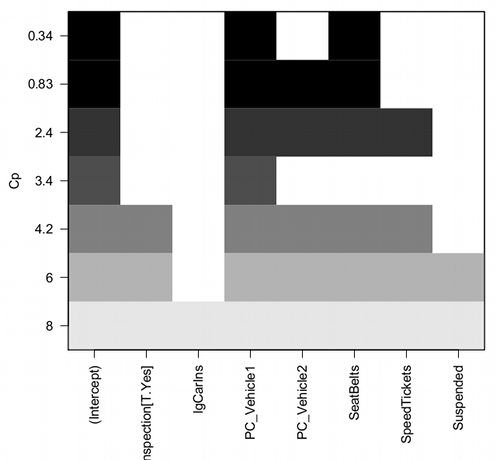
Figure 2. Mallow’s Cp plot
Lets break down the plot. First define the axes. The vertical axis is the range of values for the Cp calculated for each model. The horizontal axis is categorical and reads from left to right: Intercept, Inspection[T.Yes], etc., up to Suspended. Looking into the graph itself we see horizontal bars — the extent of shading indicates which model corresponds to the Cp value. For example, the lowest bar which is associated with the Cp value of 8 extends all the way to the right of the graph. This says that the model evaluated included all of the variables and therefore was the saturated or full model. The next bar from the bottom of the graph is missing only one block (lgCarIns), which tells us the Cp value 6 corresponds to a reduced model, and so forth.
Cross-validation
Once you have identified your Best Fit model, then, your proceed to run the diagnostics plots. For the rest of the discussion we return to our first example.
Rcmdr: Models → Graphs → Basic diagnostic plots.
We’ll just concern ourselves with the first row of plots (Fig. 3).
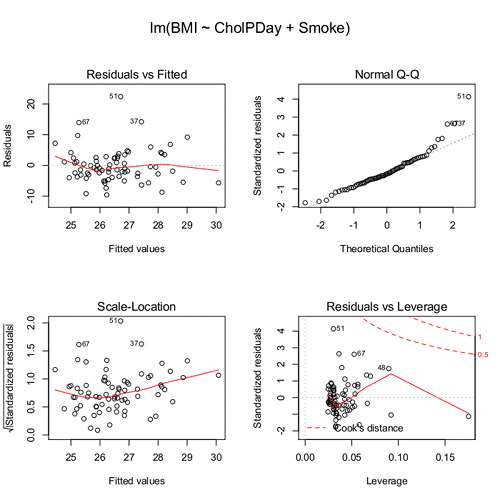
Figure 3. Diagnostic plots
The left one shows the residuals versus the predicted values — if you see a trend here, the assumption of linearity has been violated. The second plot is a test of the assumption of normality of the residuals. Interpret them (residuals OK, Residuals normally distributed? Yes/ No), and you’re done. Here, I would say I see no real trend in the residuals vs fitted plot, so assumption of linear fit is OK. For normality, there is a tailing off at the larger values of residuals, which might be of some concern (and I would start thinking about possible leverage problems), but nothing dramatic. I would conclude that our Model 6 is a good fitting model and one that could be used to make predictions.
Now, if you think a moment, you should identify a logical problem. We used the same data to “check” the model fit as we did to make the model in the first place. In particular if the model is intended to make predictions it would be advisable to check the performance of the model (e.g., does it make reasonable predictions?) by supplying new data, data not used to construct the model, into the model. If new data are not available, one acceptable practice is to divide the full data set into at least two subsets, one used to develop the model (sometimes called the calibration or training dataset) and the other used to test the model. The benefits of cross-validation include testing for influence points, over fitting of model parameters, and a reality check on the predictions generated from the model.
A note on best practices
In the 1980s, researchers often “removed” the effect of a covariate by first fitting a regression and then saving the residuals to use in separate analyses, such as t-tests, ANOVAs, or variance component estimates (eg, Dohm et al 2001). For example, in a study of plant growth, scientists might regress final plant height on initial size to remove the effect of size, then compare the residuals between fertilizer groups. While this approach adjusts for the covariate, it is problematic because residuals are not raw data: they are correlated, have constrained sums, and underestimate uncertainty. This can distort group differences, especially if the covariate distributions differ among groups, and it ignores the variability introduced by the first regression. This approach was common in ecology, evolutionary biology, physiology, and psychology before modern mixed modeling and generalized regression workflows became standard.
Modern best practices instead recommend fitting a single unified model that includes both the covariate and the grouping variable directly. Using the same plant growth example, a linear model such as  estimates the effect of fertilizer while properly accounting for initial plant size. This approach provides valid standard errors, allows correct inference about group differences, and can accommodate interactions (eg, if the relationship between
estimates the effect of fertilizer while properly accounting for initial plant size. This approach provides valid standard errors, allows correct inference about group differences, and can accommodate interactions (eg, if the relationship between  and
and  differs by group) or hierarchical data structures using mixed-effects models. Additionally, modern workflows include diagnostics such as residual plots, Cook’s Distance, and variance inflation factors to check model assumptions and influential points. Use mixed models when data are hierarchical or repeated data. Random effects handle variation among groups, individuals, years, etc., in a single coherent model. When covariates need to be “controlled for,” we use partial regression. This happens inside the model, not by subtracting covariate effects first. By analyzing all relevant predictors in a single model, researchers obtain more accurate estimates, properly reflect uncertainty, and avoid the statistical pitfalls of the old residual-based two-stage approach.
differs by group) or hierarchical data structures using mixed-effects models. Additionally, modern workflows include diagnostics such as residual plots, Cook’s Distance, and variance inflation factors to check model assumptions and influential points. Use mixed models when data are hierarchical or repeated data. Random effects handle variation among groups, individuals, years, etc., in a single coherent model. When covariates need to be “controlled for,” we use partial regression. This happens inside the model, not by subtracting covariate effects first. By analyzing all relevant predictors in a single model, researchers obtain more accurate estimates, properly reflect uncertainty, and avoid the statistical pitfalls of the old residual-based two-stage approach.
Thus, there are several statistical reasons the older approach fails under modern scrutiny. First, Residuals are not independent observations. Residuals are model-dependent quantities, not raw data. Once you fit a regression, the residuals are correlated with each other, constrained to sum to zero, and, likely heteroscedastic if the model was imperfect. Using them in a new test violates standard assumptions of the second test.
Second, you underestimate uncertainty. If you perform tests on residuals, the standard errors do not include uncertainty from the first regression, which may lead to inflated Type I error and an overly confident “adjusted” group comparisons.
Third, the old approach creates a “two-stage model” that breaks the likelihood framework. A two-stage analysis discards the correct joint likelihood and therefore produces biases p-values, and interferes with valid confidence intervals or predictions.
Under the modern one-stage models include ML/REML, generalized estimating equations, and fully integrated Bayesian models, which keeps the model coherent. Use of mixed models make residualization unnecessary. In studies with repeated measures, blocks, sites, subjects, or years, the old approach cannot partition sources of variation correctly.
In short, the advantages of the one-stage approach is that resulting models have valid uncertainty, correct group comparisons, better handling of interactions and hierarchy, full likelihood-based inference. They fit a single unified regression or mixed-effects model with all relevant predictors, examine diagnostics, and make inferences directly. This is one more benefit of the increased power and availability of more powerful computers and more complete development of mixed methods modeling.
Questions
- Write up three learning outcomes for this page. Hint: Point your favorite generative AI to this page and ask for help.
Quiz Chapter 18.5
Selecting the best regression model
Chapter 18 contents
18.2 – Nonlinear regression
Introduction
The linear model is incredibly relevant in so many cases. A quick look “linear model” in PUBMED returns about 22 thousand hits; 3.7 million in Google Scholar; 3 thousand hits in ERIC database. These results compare to search of “statistics” in the same databases: 2.7 million (PUBMED), 7.8 million (Google Scholar), 61.4 thousand (ERIC). But all models are not the same.
Fit of a model to the data can be evaluated by looking at the plots of residuals (Fig 1), where we expect to find random distribution of residuals across the range of predictor variable.
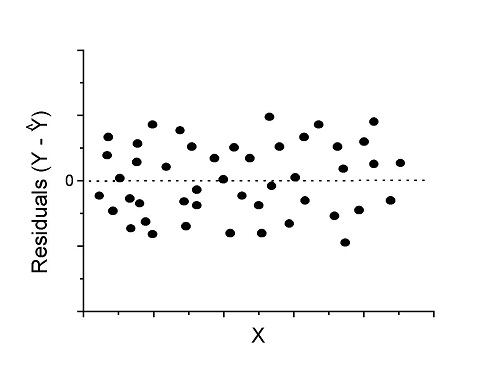
Figure 1. Ideal plot of residuals against values of X, the predictor variable, for a well-supported linear model fit to the data.
However, clearly, there are problems for which assumption of fit to line is not appropriate. We see this, again, in patterns of residuals, eg, Figure 2.
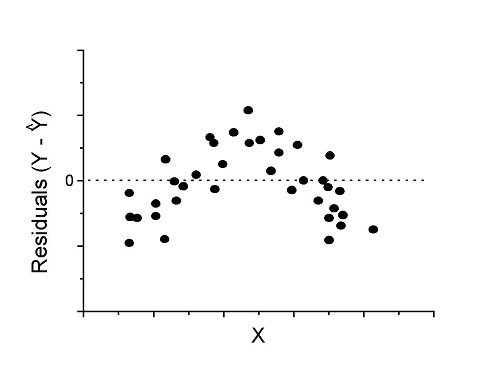
Figure 2. Example of residual plot; pattern suggests nonlinear fit.
Fitting of polynomial linear model
Fit simple linear regression, data set Yuan et al lifespan, listed below.
R code
LinearModel.1 <- lm(cumFreq~Months, data=yuan)
summary(LinearModel.1)
Call:
lm(formula = cumFreq ~ Months, data = yuan)
Residuals:
Min 1Q Median 3Q Max
-0.11070 -0.07799 -0.01728 0.06982 0.13345
Coefficients:
Estimate Std. Error t value Pr(>|t|)
(Intercept) -0.132709 0.045757 -2.90 0.0124 *
Months 0.029605 0.001854 15.97 6.37e-10 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Residual standard error: 0.09308 on 13 degrees of freedom
Multiple R-squared: 0.9515, Adjusted R-squared: 0.9477
F-statistic: 254.9 on 1 and 13 DF, p-value: 6.374e-10
We see from the R2 (95%), a high degree of fit to the data. However, residual plot reveals obvious trend (Fig 3)
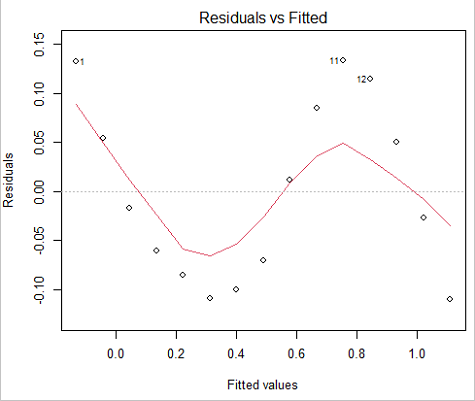
Figure 3. Residual plot, residuals against fitted values.
We can fit a polynomial regression.
First, a second order polynomial
LinearModel.2 <- lm(cumFreq ~ poly( Months, degree=2), data=yuan)
summary(LinearModel.2)
Call:
lm(formula = cumFreq ~ poly(Months, degree = 2), data = yuan)
Residuals:
Min 1Q Median 3Q Max
-0.13996 -0.06720 -0.02338 0.07153 0.14277
Coefficients:
Estimate Std. Error t value Pr(>|t|)
(Intercept) 0.48900 0.02458 19.891 1.49e-10 ***
poly(Months, degree = 2)1 1.48616 0.09521 15.609 2.46e-09 ***
poly(Months, degree = 2)2 0.06195 0.09521 0.651 0.528
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Residual standard error: 0.09521 on 12 degrees of freedom
Multiple R-squared: 0.9531, Adjusted R-squared: 0.9453
F-statistic: 122 on 2 and 12 DF, p-value: 0.0000000106
Second, try a third order polynomial
LinearModel.3 <- lm(cumFreq ~ poly(Months, degree = 3), data=yuan)
summary(LinearModel.3)
Call:
lm(formula = cumFreq ~ poly(Months, degree = 3), data = yuan)
Residuals:
Min 1Q Median 3Q Max
-0.052595 -0.021533 0.001023 0.025166 0.048270
Coefficients:
Estimate Std. Error t value Pr(>|t|)
(Intercept) 0.488995 0.008982 54.442 9.90e-15 ***
poly(Months, degree = 3)1 1.486157 0.034787 42.722 1.41e-13 ***
poly(Months, degree = 3)2 0.061955 0.034787 1.781 0.103
poly(Months, degree = 3)3 -0.308996 0.034787 -8.883 2.38e-06 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Residual standard error: 0.03479 on 11 degrees of freedom
Multiple R-squared: 0.9943, Adjusted R-squared: 0.9927
F-statistic: 635.7 on 3 and 11 DF, p-value: 1.322e-12
Which model is best? We are tempted to compare R-squared among the models, but R2 turn out to be untrustworthy here. Instead, we compare using Akaike Information Criterion, AIC
R code/results
AIC(LinearModel.1,LinearModel.2, LinearModel.3)
df AIC
LinearModel.1 3 -24.80759
LinearModel.2 4 -23.32771
LinearModel.3 5 -52.83981
Smaller the AIC, better fit.
anova(RegModel.5,LinearModel.3, LinearModel.4) Analysis of Variance Table Model 1: cumFreq ~ Months Model 2: cumFreq ~ poly(Months, degree = 2) Model 3: cumFreq ~ poly(Months, degree = 3) Res.Df RSS Df Sum of Sq F Pr(>F) 1 13 0.112628 2 12 0.108789 1 0.003838 3.1719 0.1025 3 11 0.013311 1 0.095478 78.9004 0.000002383 *** --- Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Logistic regression
The Logistic regression is a classic example of nonlinear model.
R code
logisticModel <-nls(yuan$cumFreq~DD/(1+exp(-(CC+bb*yuan$Months))), start=list(DD=1,CC=0.2,bb=.5),data=yuan,trace=TRUE)
5.163059 : 1.0 0.2 0.5
2.293604 : 0.90564552 -0.07274945 0.11721201
1.109135 : 0.96341283 -0.60471162 0.05066694
0.429202 : 1.29060000 -2.09743525 0.06785993
0.3863037 : 1.10392723 -2.14457296 0.08133307
0.2848133 : 0.9785669 -2.4341333 0.1058674
0.1080423 : 0.9646295 -3.1918526 0.1462331
0.005888491 : 1.0297915 -4.3908114 0.1982491
0.004374918 : 1.0386521 -4.6096564 0.2062024
0.004370212 : 1.0384803 -4.6264657 0.2068853
0.004370201 : 1.0385065 -4.6269276 0.2068962
0.004370201 : 1.0385041 -4.6269822 0.2068989
summary(logisticModel)
Formula: yuan$cumFreq ~ DD/(1 + exp(-(CC + bb * yuan$Months)))
Parameters:
Estimate Std. Error t value Pr(>|t|)
DD 1.038504 0.014471 71.77 < 2e-16 ***
CC -4.626982 0.175109 -26.42 5.29e-12 ***
bb 0.206899 0.008777 23.57 2.03e-11 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Residual standard error: 0.01908 on 12 degrees of freedom
Number of iterations to convergence: 11
Achieved convergence tolerance: 0.000006909
> AIC(logisticModel)
[1] -71.54679
Logistic regression is a statistical method for modeling the dependence of a categorical (binomial) outcome variable on one or more categorical and continuous predictor variables (Bewick et al 2005).
The logistic function is used to transform a sigmoidal curve to a more or less straight line while also changing the range of the data from binary (0 to 1) to infinity (-∞,+∞). For event with probability of occurring p, the logistic function is written as

where ln refers to the natural logarithm.
The logit link function is used in models to transform a probability, which is restricted between 0 and 1, into the log-odds scale, which ranges from negative infinity to positive infinity. This transformation makes the relationship between predictors and the outcome linear. In other words, this is an odds ratio. It represents the effect of the predictor variable on the chance that the event will occur. Much of our contingency table work in Chapter 7 would be best analyzed with the logistic regression.
The logistic regression model then very much resembles the same as we have seen before.

In R and Rcmdr we use the glm() function to model the logistic function. Logistic regression is used to model a binary outcome variable. What is a binary outcome variable? It is categorical! Examples include: Living or Dead; Diabetes Yes or No; Coronary artery disease Yes or No. Male or Female. One of the categories could be scored 0, the other scored 1. For example, living might be 0 and dead might be scored as 1. (By the way, for a binomial variable, the mean for the variable is simply the number of experimental units with “1” divided by the total sample size.)
With the addition of a binary response variable, we are now really close to the Generalized Linear Model. Now we can handle statistical models in which our predictor variables are either categorical or ratio scale. All of the rules of crossed, balanced, nested, blocked designs still apply because our model is still of a linear form.
We write our generalized linear model
just to distinguish it from a general linear model with the ratio-scale Y as the response variable.
Think of the logistic regression as modeling a threshold of change between the 0 and the 1 value. In another way, think of all of the processes in nature in which there is a slow increase, followed by a rapid increase once a transition point is met, only to see the rate of change slow down again. Growth is like that. We start small, stay relatively small until birth, then as we reach our early teen years, a rapid change in growth (height, weight) is typically seed (well, not in my case … at least for the height). The fitted curve I described is a logistic one (other models exist too). Where the linear regression function was used to minimize the squared residuals as the definition of the best fitting line, now we use the logistic as one possible way to describe or best fit this type of a curved relationship between an outcome and one or more predictor variables. We then set out to describe a model which captures when an event is unlikely to occur (the probability of dying is close to zero) AND to also describe when the event is highly likely to occur (the probability is close to one).
A simple way to view this is to think of time being the predictor (X) variable and risk of dying. If we’re talking about the lifetime of a mouse (lifespan typically about 18-36 months), then the risk of dying at one months is very low, and remains low through adulthood until the mouse begins the aging process. Here’s what the plot might look like, with the probability of dying at age X on the Y axis (probability = 0 to 1) (Fig 4).
Figure 4. Lifespan of 1881 mice from 31 inbred strains (Data from Yuan et al (2012) available at https://phenome.jax.org/projects/Yuan2).
We ask — of all the possible models we could draw, which best fits the data? The curve fitting process is called the logistic regression.
With some minor, but important differences, running the logistic regression is the same as what you have been doing so far for ANOVA and for linear regression. In Rcmdr, access the logistic regression function by invoking the Generalized Linear Model (Fig 5).
Rcmdr: Statistics → Fit models → Generalized linear model.
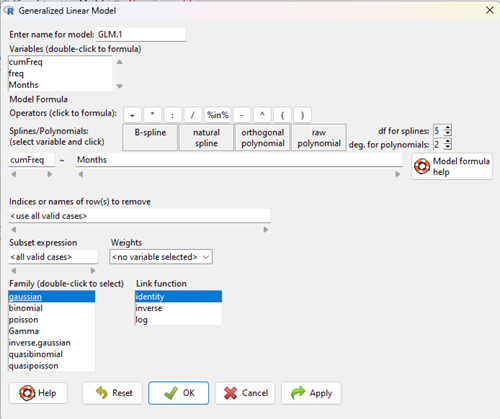
Figure 5. Screenshot Rcmdr GLM menu. For logistic on ratio-scale dependent variable, select gaussian family and identity link function.
Select the model as before. The box to the left accepts your binomial dependent variable; the box at right accepts your factors, your interactions, and your covariates. It permits you to inform R how to handle the factors: Crossed? Just enter the factors and follow each with a plus. If fully crossed, then the interactions may be specified with “:” to explicitly call for a two-way interaction between two (A:B) or a three-way interaction between three (A:B:C) variables. In the later case, if all of the two way interactions are of interest, simply typing A*B*C would have done it. If nested, then use %in% to specify the nesting factor.
R output
> GLM.1 <- glm(cumFreq ~ Months, family=gaussian(identity), data=yuan) > summary(GLM.1) Call: glm(formula = cumFreq ~ Months, family = gaussian(identity), data = yuan) Deviance Residuals: Min 1Q Median 3Q Max -0.11070 -0.07799 -0.01728 0.06982 0.13345 Coefficients: Estimate Std. Error t value Pr(>|t|) (Intercept) -0.132709 0.045757 -2.90 0.0124 * Months 0.029605 0.001854 15.97 6.37e-10 *** --- Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1 (Dispersion parameter for gaussian family taken to be 0.008663679) Null deviance: 2.32129 on 14 degrees of freedom Residual deviance: 0.11263 on 13 degrees of freedom AIC: -24.808 Number of Fisher Scoring iterations: 2
Assessing fit of the logistic regression model
Some of the differences you will see with the logistic regression is the term deviance. Deviance in statistics simply means compare one model to another and calculate some test statistic we’ll call “the deviance.” We then evaluate the size of the deviance like a chi-square goodness of fit. If the model fits the data poorly (residuals large relative to the predicted curve), then the deviance will be small and the probability will also be high — the model explains little of the data variation. On the other hand, if the deviance is large, then the probability will be small — the model explains the data, and the probability associated with the deviance will be small (significantly so? You guessed it! P < 0.05).
The Wald test statistic
where n and β refers to any of the n coefficient from the logistic regression equation and SE refers to the standard error if the coefficient. The Wald test is used to test the statistical significance of the coefficients. It is distributed approximately as a chi-squared probability distribution with one degree of freedom. The Wald test is reasonable, but has been found to give values that are not possible for the parameter (eg, negative probability).
Likelihood ratio tests are generally preferred over the Wald test. For a coefficient, the likelihood test is written as
![]()
where L0 is the likelihood of the data when the coefficient is removed from the model (ie, set to zero value), whereas L1 is the likelihood of the data when the coefficient is the estimated value of the coefficient. It is also distributed approximately as a chi-squared probability distribution with one degree of freedom.
Questions
- Write up three learning outcomes for this page. Hint: Point your favorite generative AI to this page and ask for help.
- What is the primary difference between linear regression and logistic regression?
- What type of dependent variable is used in logistic regression?
- What is the purpose of the sigmoid (or logistic) function in this model?
- What is the role of a significance test (eg, a Wald test) in logistic regression?
- Why is the LRT generally considered more reliable than the Wald test for logistic regression, especially with smaller sample sizes?
Quiz Chapter 18.2
Nonlinear regression
Data set
Yuan data
| Months | freq | cumFreq |
|---|---|---|
| 0 | 0 | 0 |
| 3 | 0.010633 | 0.010633 |
| 6 | 0.017012 | 0.027645 |
| 9 | 0.045189 | 0.072834 |
| 12 | 0.064327 | 0.137161 |
| 15 | 0.064859 | 0.20202 |
| 18 | 0.09782 | 0.299841 |
| 21 | 0.118554 | 0.418394 |
| 24 | 0.171186 | 0.58958 |
| 27 | 0.162148 | 0.751728 |
| 30 | 0.137161 | 0.888889 |
| 33 | 0.069644 | 0.958533 |
| 36 | 0.024455 | 0.982988 |
| 39 | 0.011696 | 0.994684 |
| 42 | 0.005316 | 1 |